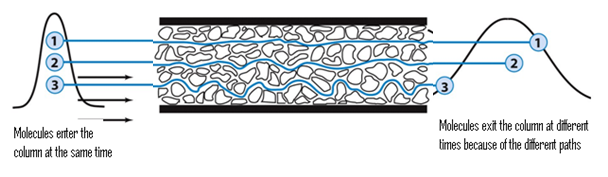
 adsorptievermogen van een bepaalde stof aan een stationaire fase. Een veel gebruikte analogie is die van een winkelstraat vol kledingwinkels: een grote groep mensen staat aan het begin van een winkelstraat, zij moeten de straat doorlopen. De kledingliefhebbers doe er veel langer over dan de niet-kledingliefhebbers omdat de eerste steeds de kledingwinkels binnen gaan.
adsorptievermogen van een bepaalde stof aan een stationaire fase. Een veel gebruikte analogie is die van een winkelstraat vol kledingwinkels: een grote groep mensen staat aan het begin van een winkelstraat, zij moeten de straat doorlopen. De kledingliefhebbers doe er veel langer over dan de niet-kledingliefhebbers omdat de eerste steeds de kledingwinkels binnen gaan. papierchromatografie. De mobiele fase is dan vaak water, de stationaire fase is een stuk filtreerpapier. Door stiftpunten op het papier aan te brengen en deze in het water te zetten kan er een scheiding optreden tussen de diverse kleuren waaruit de inkt is opgebouwd.
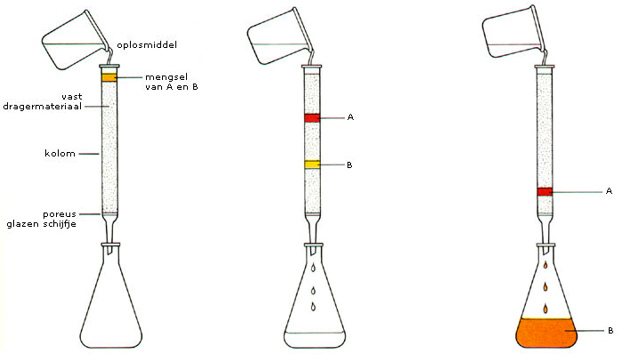
papierchromatografie. De mobiele fase is dan vaak water, de stationaire fase is een stuk filtreerpapier. Door stiftpunten op het papier aan te brengen en deze in het water te zetten kan er een scheiding optreden tussen de diverse kleuren waaruit de inkt is opgebouwd.  In deze zin is papierchromatografie vooral bedoeld als analysemethode, je kunt zien uit welke kleuren de inkt is opgebouwd.
In deze zin is papierchromatografie vooral bedoeld als analysemethode, je kunt zien uit welke kleuren de inkt is opgebouwd. 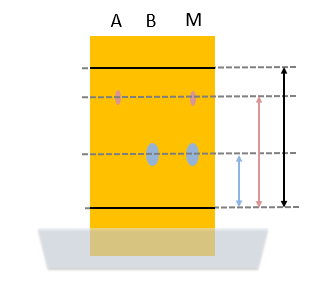 In de afbeelding rechts zie je een voorbeeld. Stof A, de rode kleurstof heeft een afstand afgelegd die overeenkomt met de rode pijl. De zwarte pijl is de afstand die het vloeistoffront heeft afgelegd vanaf de lijn waarop de monsters zijn aangebracht. De retentiefactor is dan:
In de afbeelding rechts zie je een voorbeeld. Stof A, de rode kleurstof heeft een afstand afgelegd die overeenkomt met de rode pijl. De zwarte pijl is de afstand die het vloeistoffront heeft afgelegd vanaf de lijn waarop de monsters zijn aangebracht. De retentiefactor is dan:
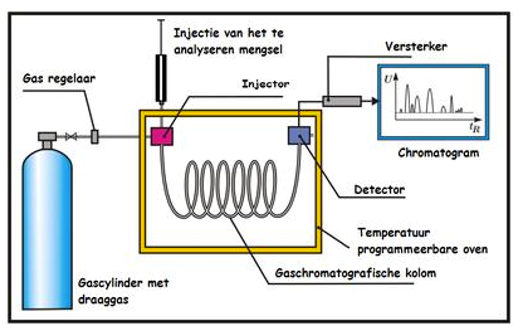
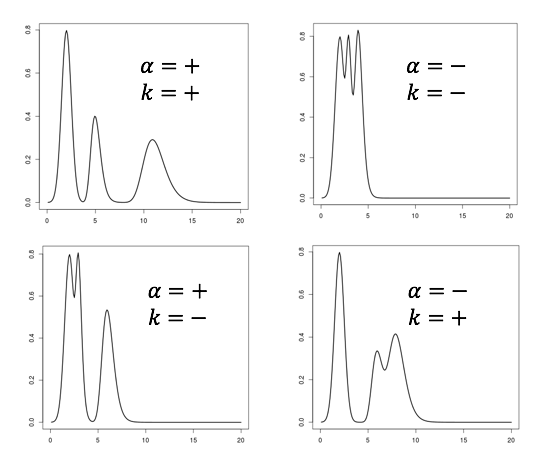
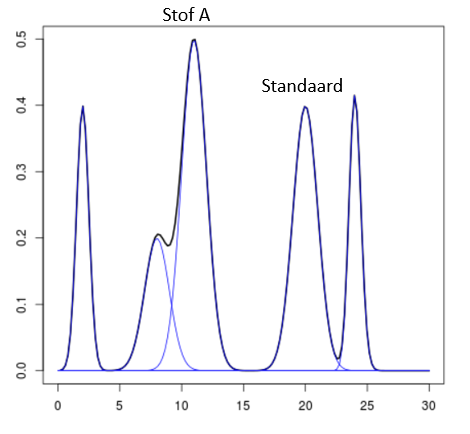 Een monster bevat een onbekende stof A die gemeten moet worden. Het chromatogram van het monster ziet er uit zoals hier rechts. Stof A heeft een retentietijd van 11 minuten. Bij het meten van het monster is 1·10–3 M van een stof S (de interne standaard) toegevoegd. De standaard heeft een retentietijd van 20 minuten. De verhouding tussen de piekoppervlakten van stof A en stof S is 1,25 : 1,00.
Een monster bevat een onbekende stof A die gemeten moet worden. Het chromatogram van het monster ziet er uit zoals hier rechts. Stof A heeft een retentietijd van 11 minuten. Bij het meten van het monster is 1·10–3 M van een stof S (de interne standaard) toegevoegd. De standaard heeft een retentietijd van 20 minuten. De verhouding tussen de piekoppervlakten van stof A en stof S is 1,25 : 1,00.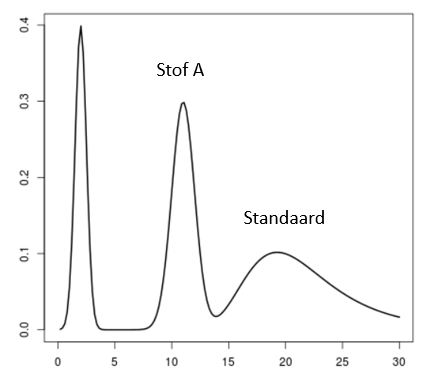 De concentraties van beide stoffen zijn gelijk aan elkaar: namelijk 1,0·10–2 M. De verhouding tussen de pieken is in dit geval 1,00 : 1,50.
De concentraties van beide stoffen zijn gelijk aan elkaar: namelijk 1,0·10–2 M. De verhouding tussen de pieken is in dit geval 1,00 : 1,50.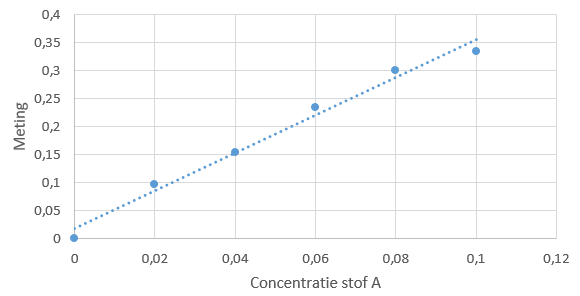 Een simpele ijklijn kan er als volgt uitzien:
Een simpele ijklijn kan er als volgt uitzien: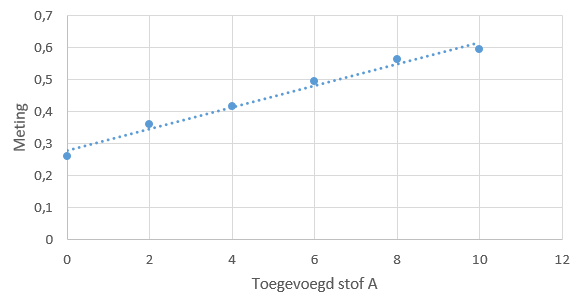 ijkreeks ziet er dan bijvoorbeeld als volgt uit:
ijkreeks ziet er dan bijvoorbeeld als volgt uit: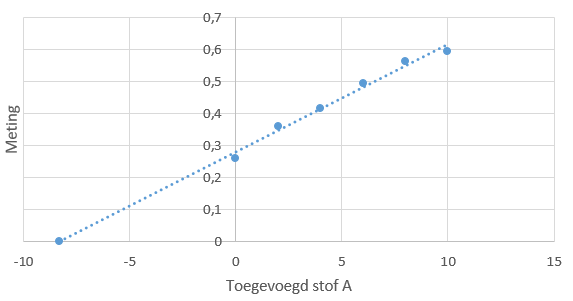

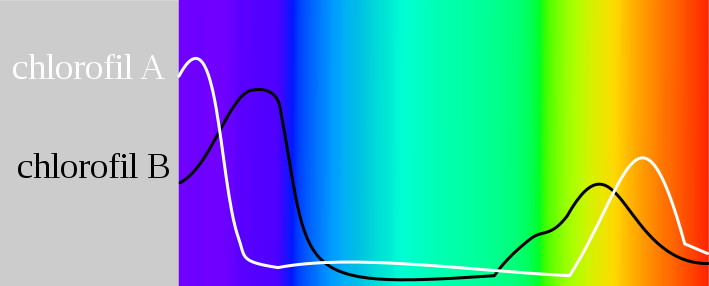 Hiernaast is duidelijk te zien dat chlorofiel vooral absorbeert in het blauwe en rode gebied. Dit zorgt ervoor dat de stoffen groen licht weerkaatsen (en wij de stof dus waarnemen als groen.
Hiernaast is duidelijk te zien dat chlorofiel vooral absorbeert in het blauwe en rode gebied. Dit zorgt ervoor dat de stoffen groen licht weerkaatsen (en wij de stof dus waarnemen als groen.
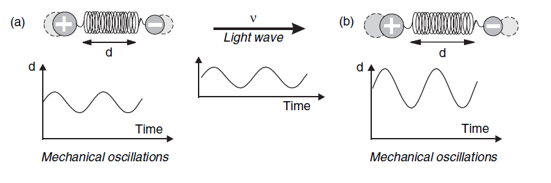
 De wet van Hooke zegt dat de mate van uitrekking van een veer proportioneel is met de kracht die daar voor nodig is:
De wet van Hooke zegt dat de mate van uitrekking van een veer proportioneel is met de kracht die daar voor nodig is: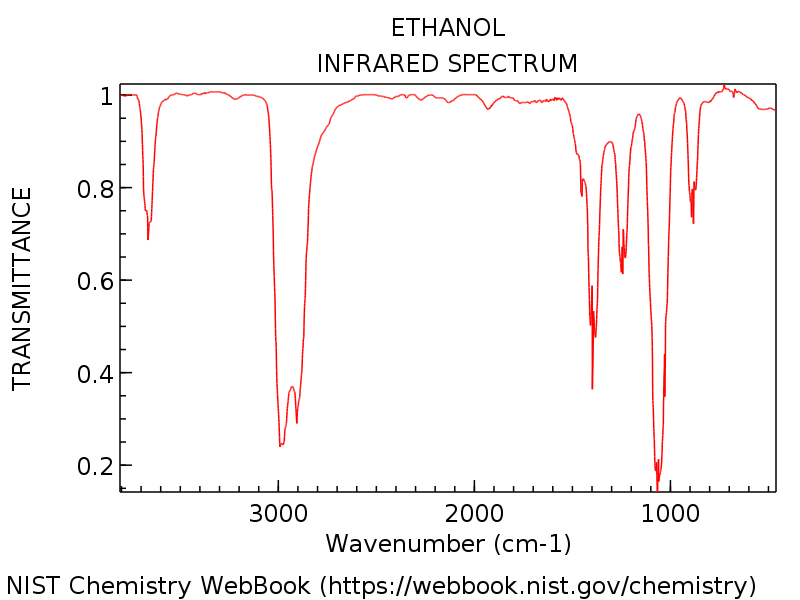
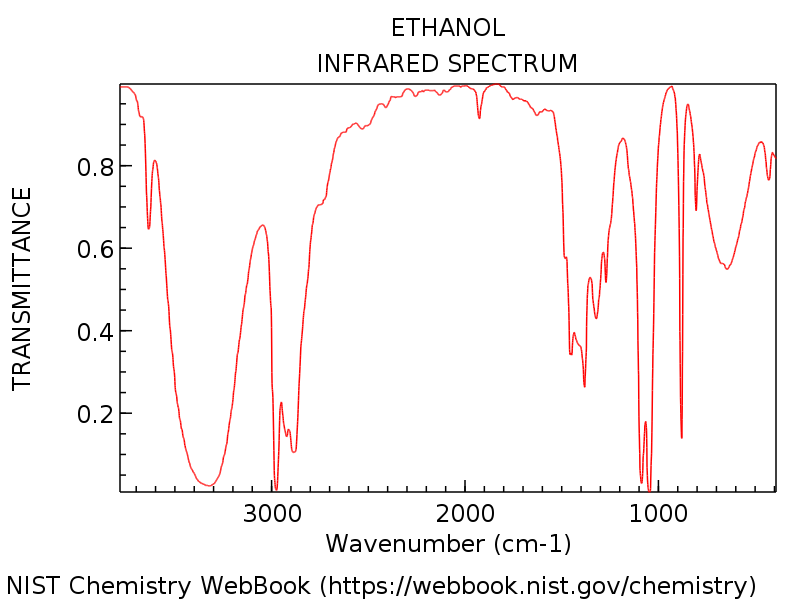 Hiernaast is nogmaals het spectrum van ethanol weergegeven, nu is het ethanolmolecuul echter niet in de gasfase (zoals hierboven het geval is) maar in de oplossing gemeten.
Hiernaast is nogmaals het spectrum van ethanol weergegeven, nu is het ethanolmolecuul echter niet in de gasfase (zoals hierboven het geval is) maar in de oplossing gemeten.
 otopen waarbij dit het geval is zijn 1H, 13C, 19F en 31P. De waterstofisotoop is de meest voorkomende en komt in vrijwel alle organische verbindingen voor. We zullen ons daarom focussen op de waterstofkernen.
otopen waarbij dit het geval is zijn 1H, 13C, 19F en 31P. De waterstofisotoop is de meest voorkomende en komt in vrijwel alle organische verbindingen voor. We zullen ons daarom focussen op de waterstofkernen.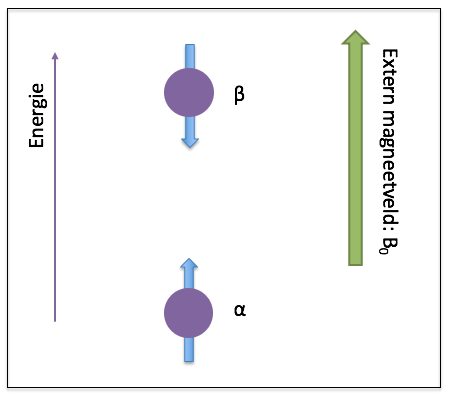
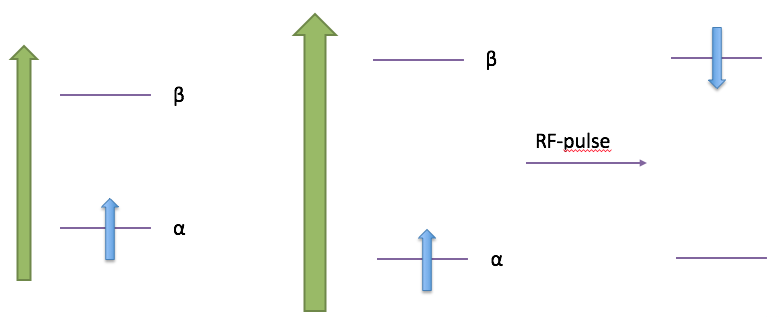
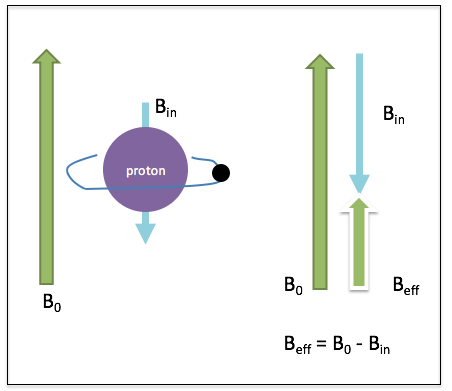
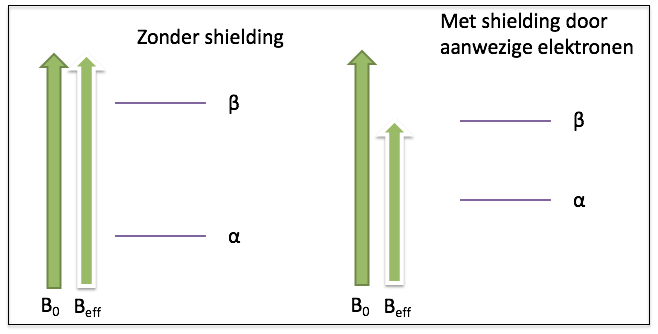
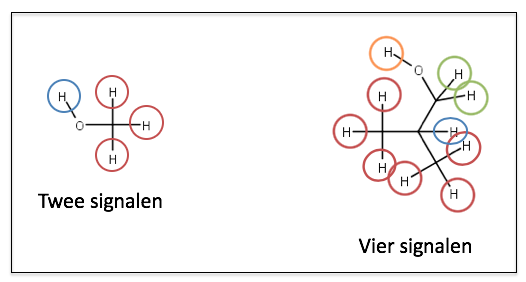
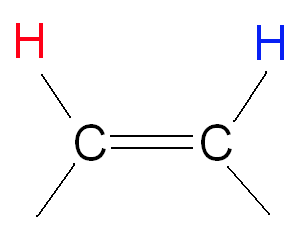
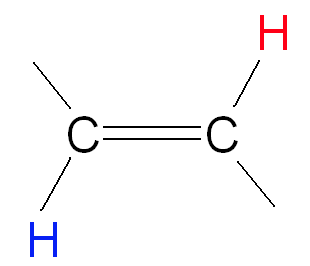
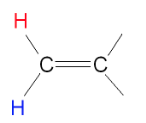
 ptisch actieve groep zitten niet equivalent. De CH2 waterstofatomen in fenylalanine zitten net naast het optisch actieve koolstofatoom. Hierdoor leveren ze in theorie een ander signaal op. In de praktijk zal dit een zeer vergelijkbaar signaal zijn wat nauwelijks te onderscheiden is.
ptisch actieve groep zitten niet equivalent. De CH2 waterstofatomen in fenylalanine zitten net naast het optisch actieve koolstofatoom. Hierdoor leveren ze in theorie een ander signaal op. In de praktijk zal dit een zeer vergelijkbaar signaal zijn wat nauwelijks te onderscheiden is.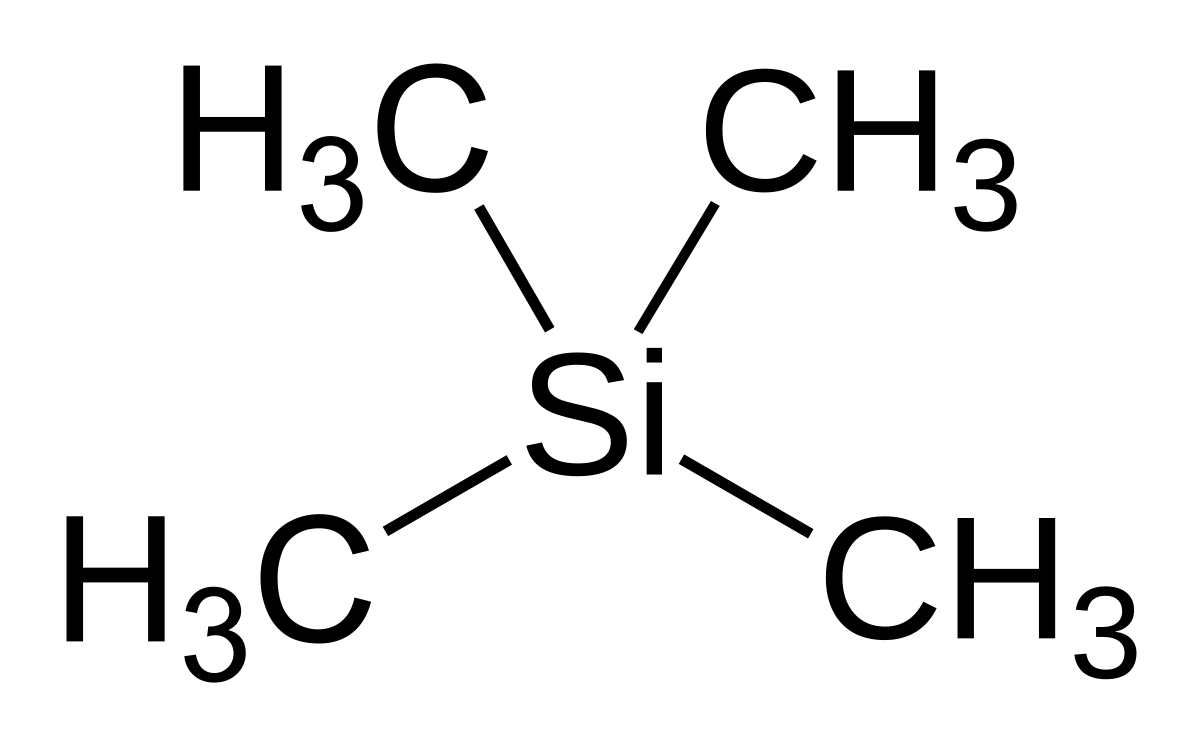 De referentiestof is tetramethylsilaan, TMS (zie rechts). Dit molecuul geeft één signaal in een spectrum. Het elektropositieve silicium atoom stuwt elektronen in de richting van de methylgroepen. Hierdoor worden de waterstofkernen in grote mate voorzien van shielding. Een grote shielding levert een klein energieverschil in spin-toestanden op en daardoor kunnen we verwachten dat het signaal een relatief lage frequentie oplevert.
De referentiestof is tetramethylsilaan, TMS (zie rechts). Dit molecuul geeft één signaal in een spectrum. Het elektropositieve silicium atoom stuwt elektronen in de richting van de methylgroepen. Hierdoor worden de waterstofkernen in grote mate voorzien van shielding. Een grote shielding levert een klein energieverschil in spin-toestanden op en daardoor kunnen we verwachten dat het signaal een relatief lage frequentie oplevert.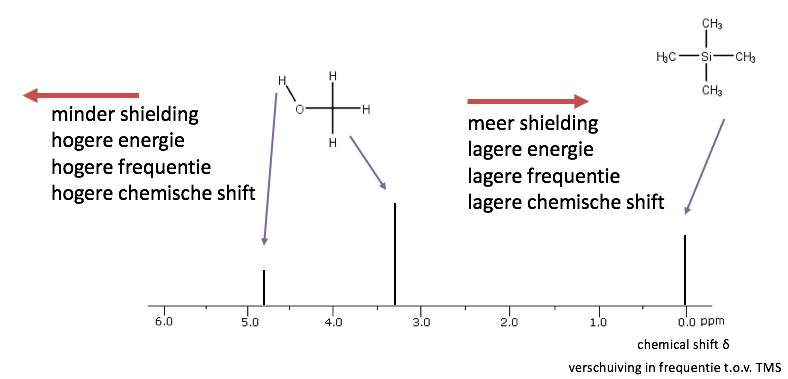
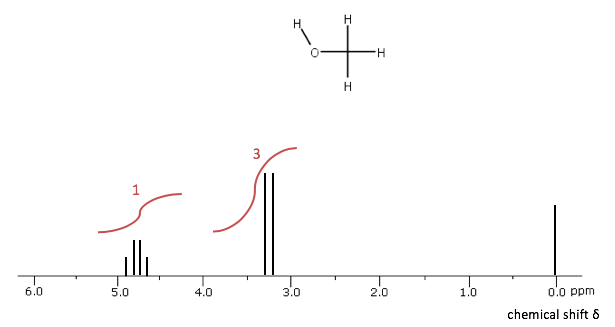
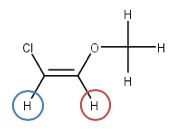 We nemen als voorbeeld de verbinding hiernaast. Het linkse blauwe proton voelt ook de effecten van het magnetische moment van het rechtse rode proton. Er zijn dan twee opties:
We nemen als voorbeeld de verbinding hiernaast. Het linkse blauwe proton voelt ook de effecten van het magnetische moment van het rechtse rode proton. Er zijn dan twee opties: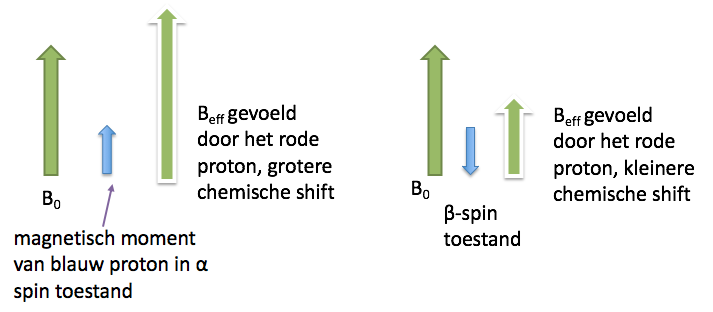
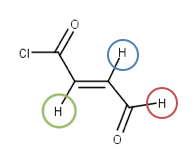 Sommige verbindingen leveren complexe splitsing van pieken op. We nemen de verbinding hier rechts als voorbeeld. Deze verbinding levert drie signalen op voor ieder van de drie aanwezige protonen. Het linkse en rechtse proton leveren allebei een doublet op, waarbij het rechtse (rode) proton minder shielding ervaart dan het linkse (groene) proton. Minder shielding wil zeggen een groter verschil tussen de spintoestanden en een grotere chemische shift. Het rode proton zal dus een doublet (vanwege het middelste - blauwe - proton als buurman) met een grote chemische shift opleveren.
Sommige verbindingen leveren complexe splitsing van pieken op. We nemen de verbinding hier rechts als voorbeeld. Deze verbinding levert drie signalen op voor ieder van de drie aanwezige protonen. Het linkse en rechtse proton leveren allebei een doublet op, waarbij het rechtse (rode) proton minder shielding ervaart dan het linkse (groene) proton. Minder shielding wil zeggen een groter verschil tussen de spintoestanden en een grotere chemische shift. Het rode proton zal dus een doublet (vanwege het middelste - blauwe - proton als buurman) met een grote chemische shift opleveren. 

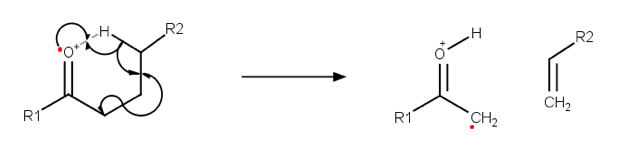
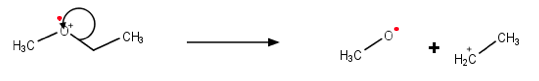
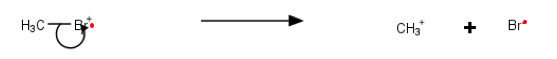
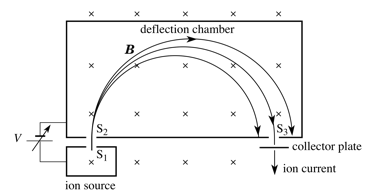 Na ionisatie en fragmentatie worden de positief geladen fragmenten versneld in een elektrisch veld. Vervolgens komen de deeltjes in een magnetisch veld waardoor deze een Lorentz-kracht ondervinden. De afbuiging van deze deeltjes is afhankelijk van de massa (en snelheid en lading) van deze deeltjes.
Na ionisatie en fragmentatie worden de positief geladen fragmenten versneld in een elektrisch veld. Vervolgens komen de deeltjes in een magnetisch veld waardoor deze een Lorentz-kracht ondervinden. De afbuiging van deze deeltjes is afhankelijk van de massa (en snelheid en lading) van deze deeltjes. 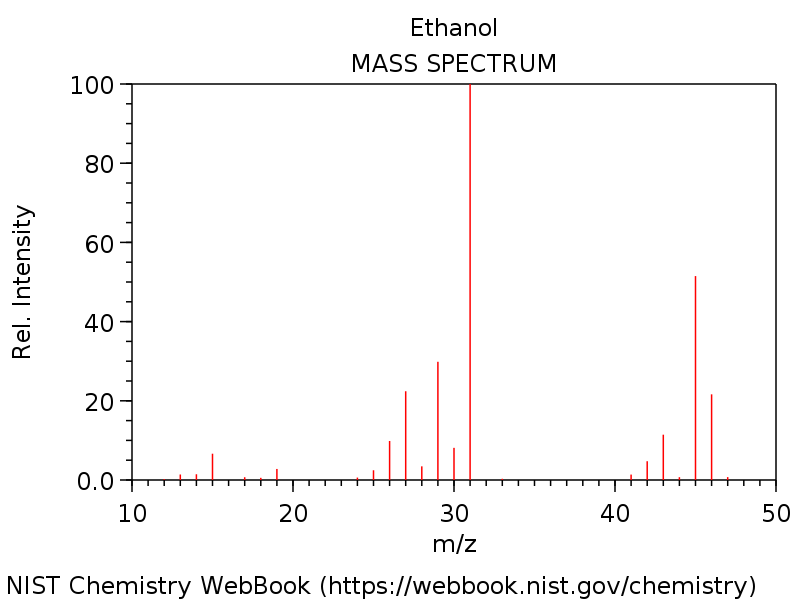

Het arrangement Life Science Instrumentele Analyse is gemaakt met Wikiwijs van Kennisnet. Wikiwijs is hét onderwijsplatform waar je leermiddelen zoekt, maakt en deelt.
- Auteur
- Laatst gewijzigd
- 27-08-2021 09:34:06
- Licentie
-


Dit lesmateriaal is gepubliceerd onder de Creative Commons Naamsvermelding 4.0 Internationale licentie. Dit houdt in dat je onder de voorwaarde van naamsvermelding vrij bent om:
- het werk te delen - te kopiëren, te verspreiden en door te geven via elk medium of bestandsformaat
- het werk te bewerken - te remixen, te veranderen en afgeleide werken te maken
- voor alle doeleinden, inclusief commerciële doeleinden.
Meer informatie over de CC Naamsvermelding 4.0 Internationale licentie.
Aanvullende informatie over dit lesmateriaal
Van dit lesmateriaal is de volgende aanvullende informatie beschikbaar:
- Toelichting
- Voor het vak Life Science ondersteunt deze wikewijs de theorie voor het voor het onderdeel analytische chemie. De werking van de diverse analyse technieken worden uitgelegd. Deze wiki kan zelfstandig gebruikt worden.
- Eindgebruiker
- leerling/student
- Moeilijkheidsgraad
- gemiddeld
- Studiebelasting
- 4 uur 0 minuten
Bronnen
| Bron | Type |
|---|---|
|
Papierchromatografie https://www.youtube.com/watch?v=uOhefwQBAbI |
Video |
|
Dunnelaagchromatografie https://www.youtube.com/watch?v=-theAxYWO2o |
Video |
|
Animatie kolomchromatografie https://www.youtube.com/watch?v=9GiLjH9Oym8 |
Video |
|
HAN - storten van een kolom https://www.youtube.com/watch?v=7CnfbTOn2Cc |
Video |
|
Vibraties ammoniak https://www.youtube.com/watch?v=aSiJ2bt1jwQ |
Video |
|
http://nmrdb.org http://nmrdb.org |
Link |
|
Extra uitleg over NMR https://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/Spectrpy/nmr/nmr1.htm |
Link |
Gebruikte Wikiwijs Arrangementen
van der Vaart, Davy. (z.d.).
Instrumentele Analyse