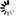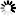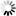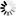




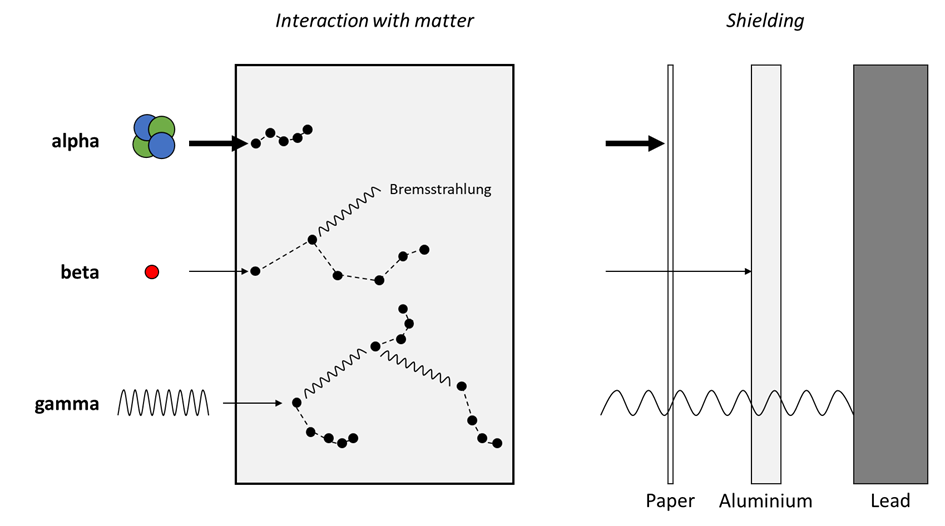



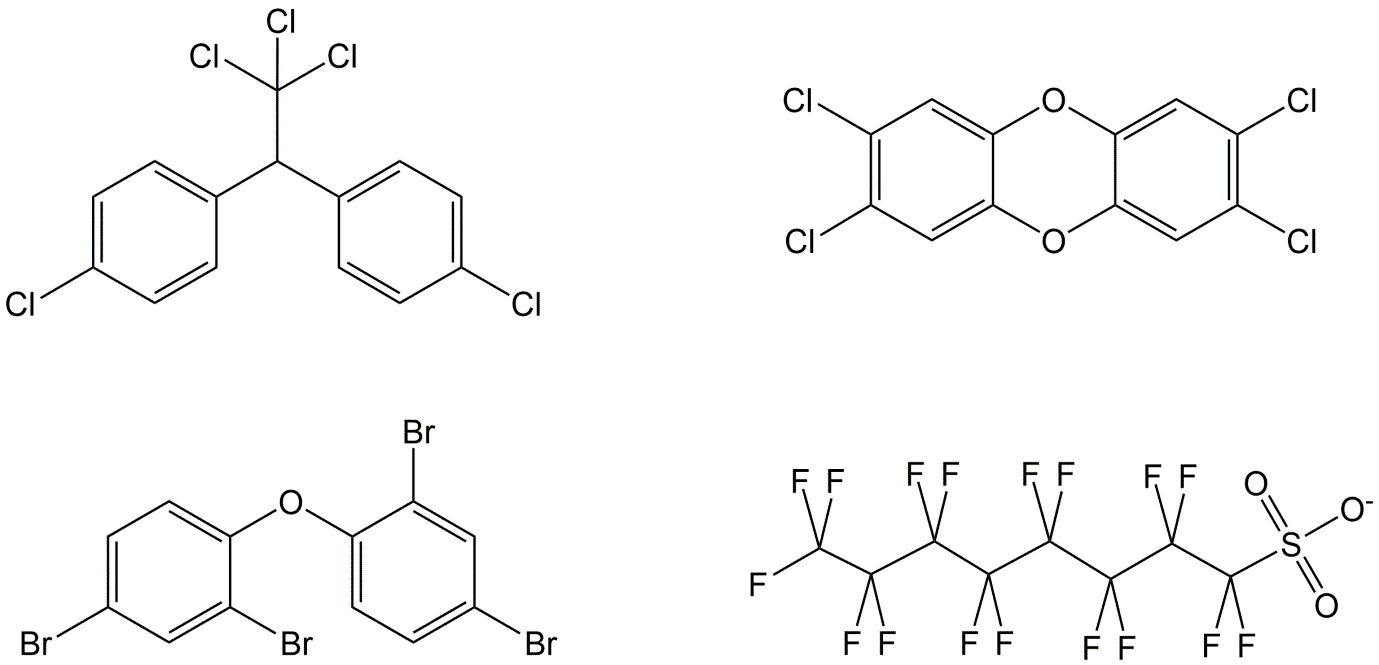


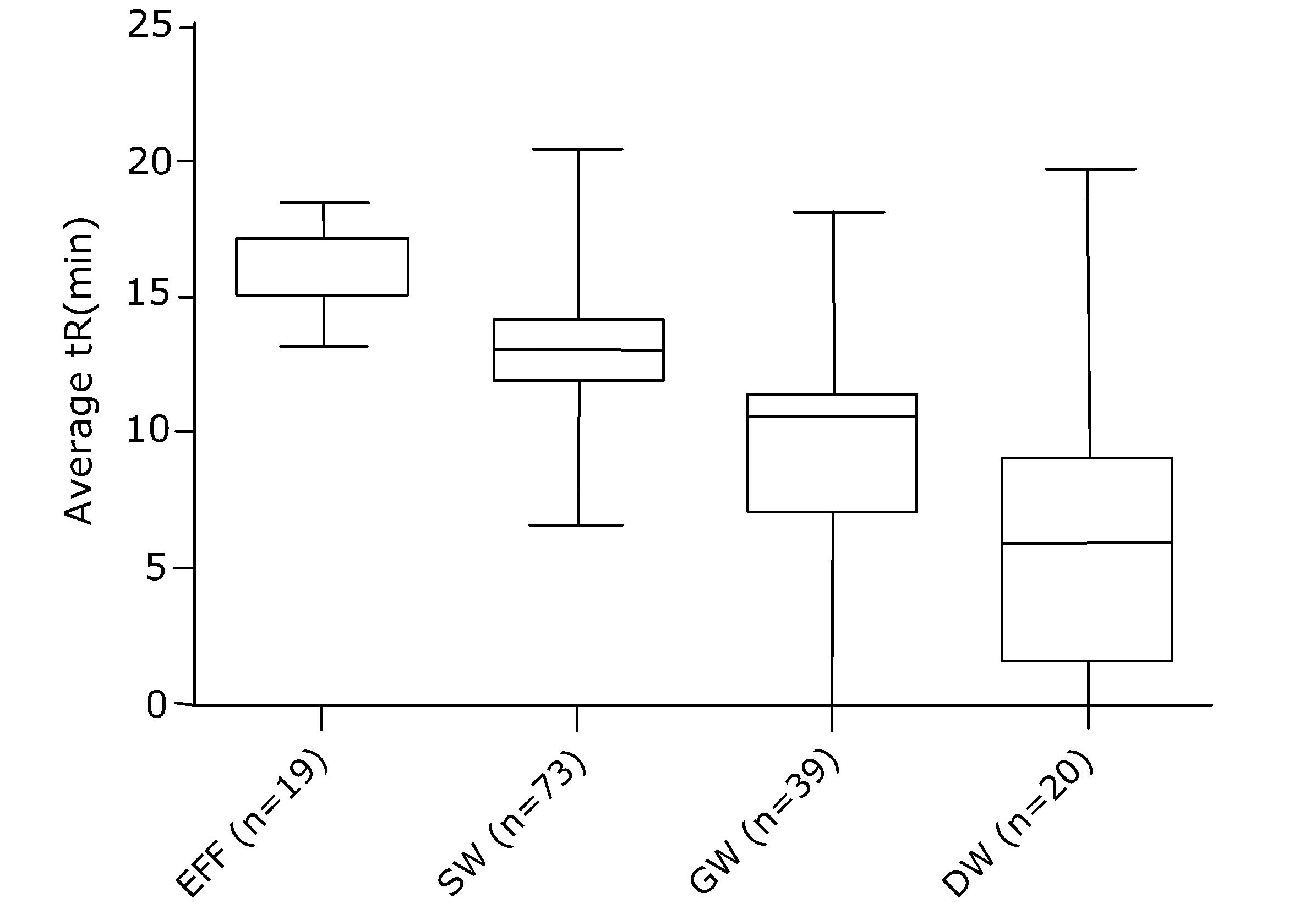

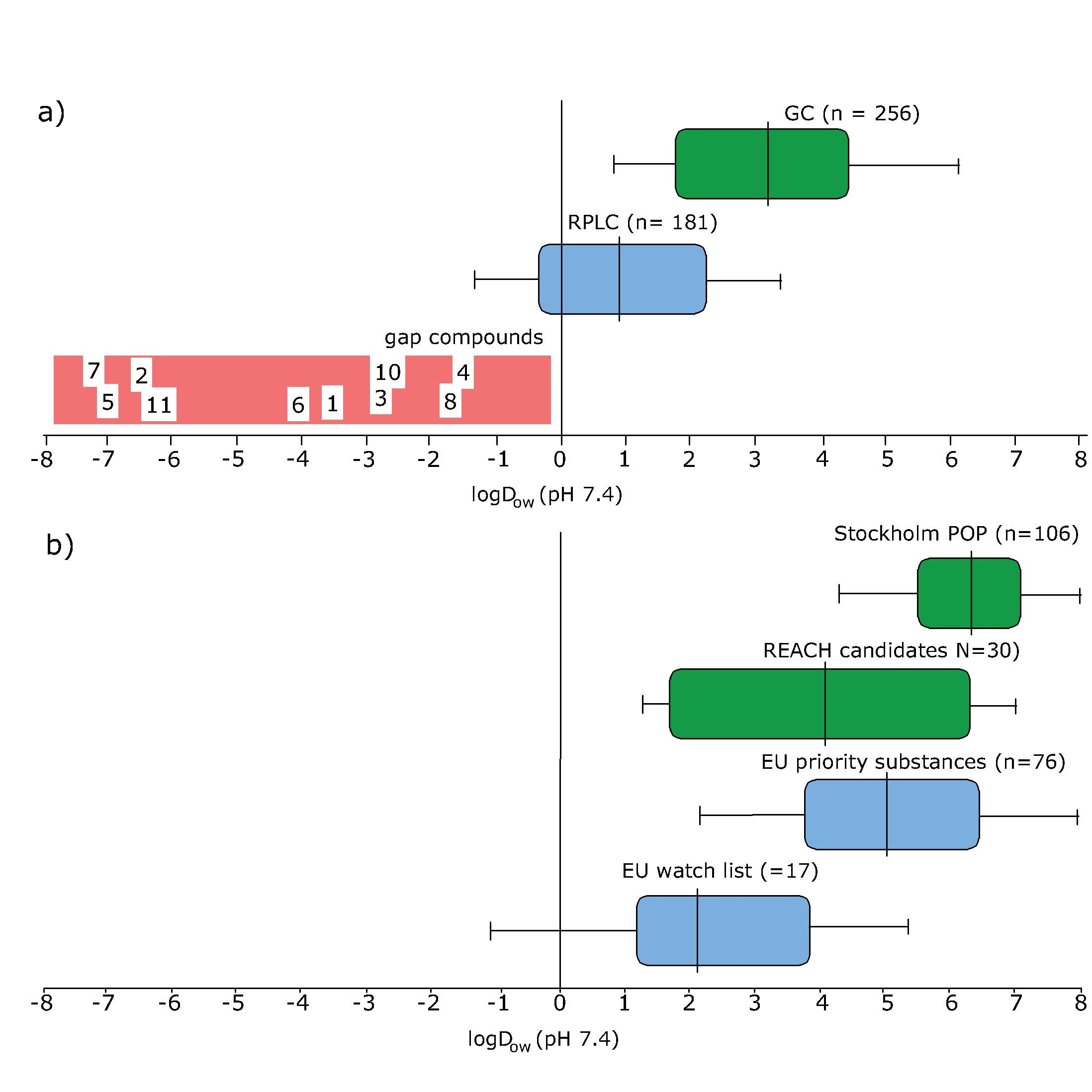
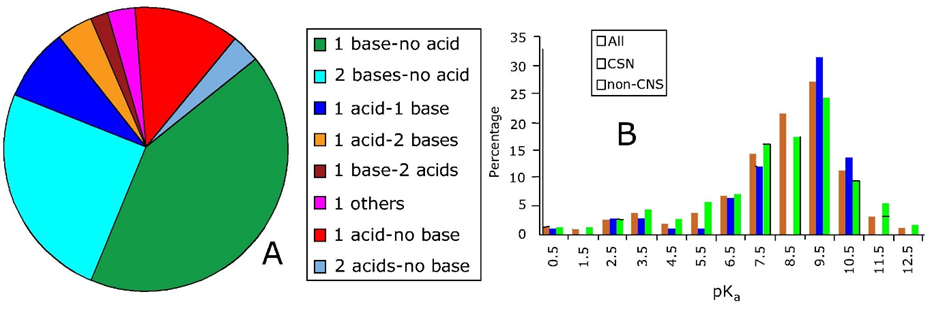


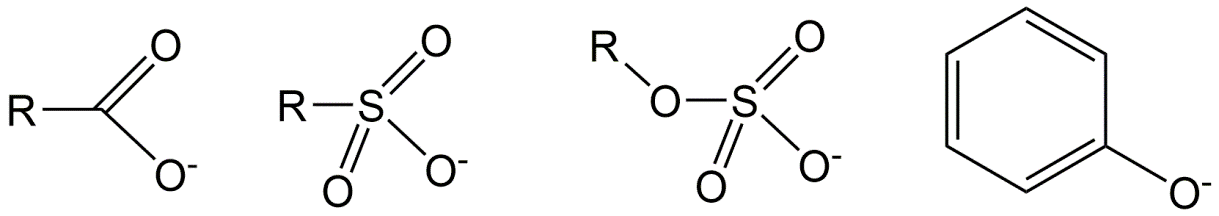
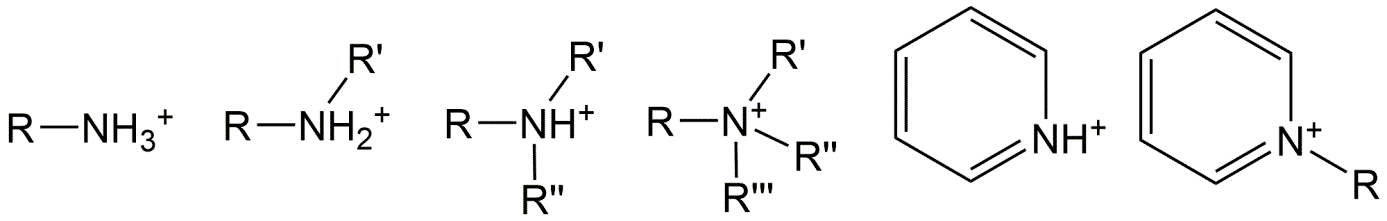
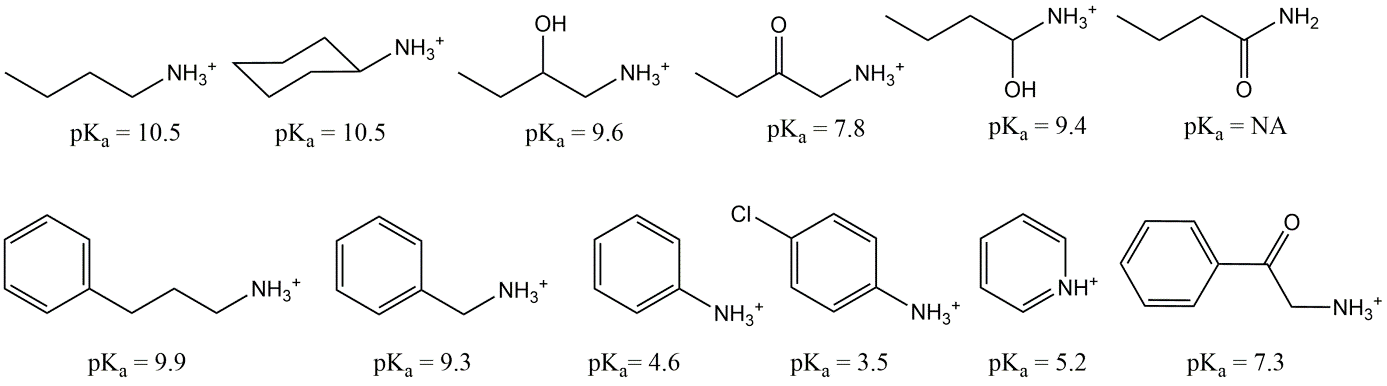







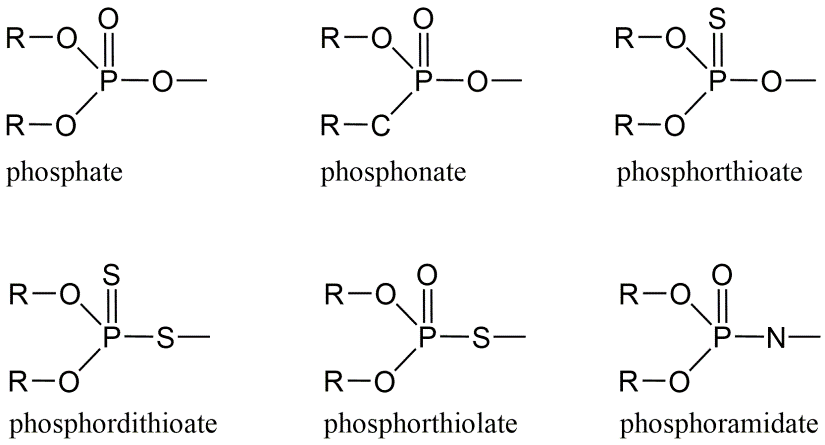

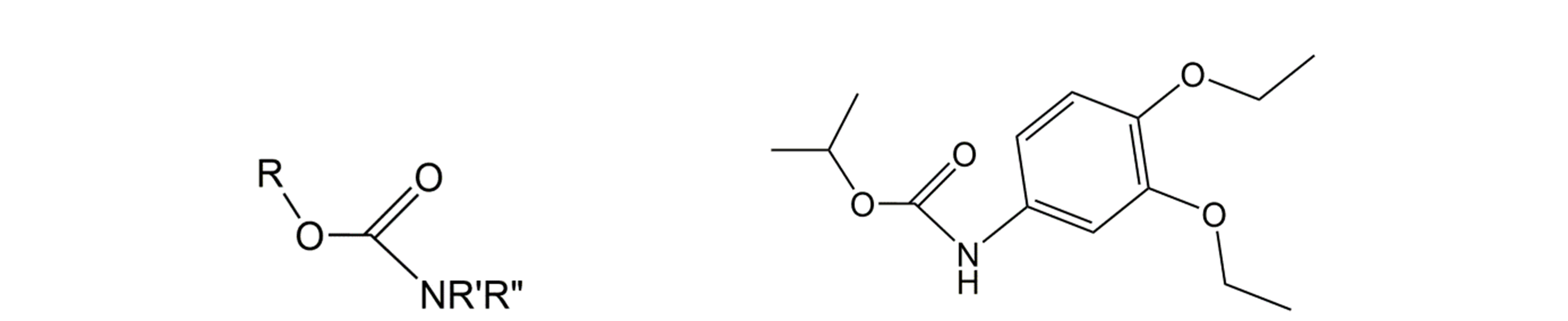



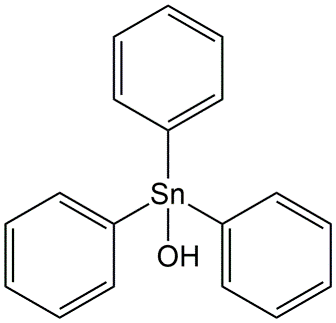
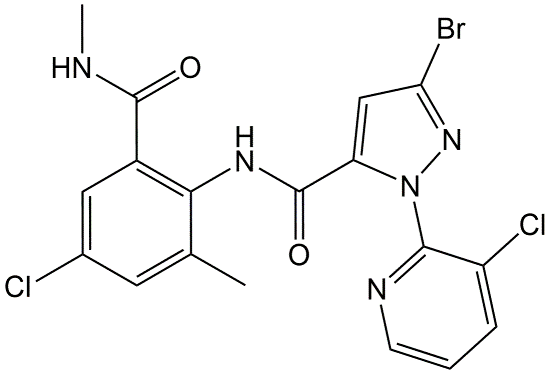
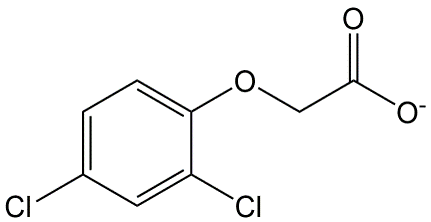
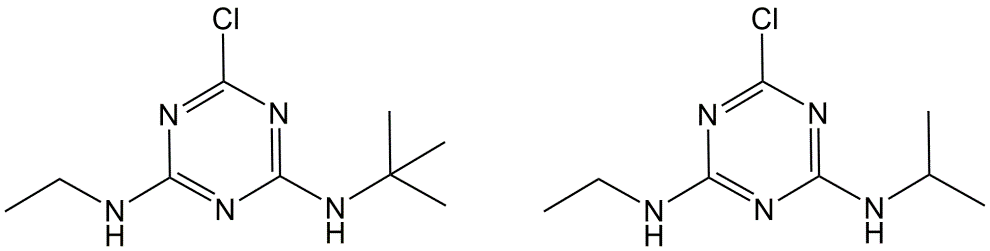
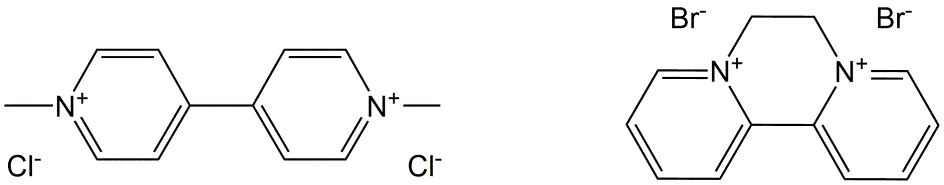
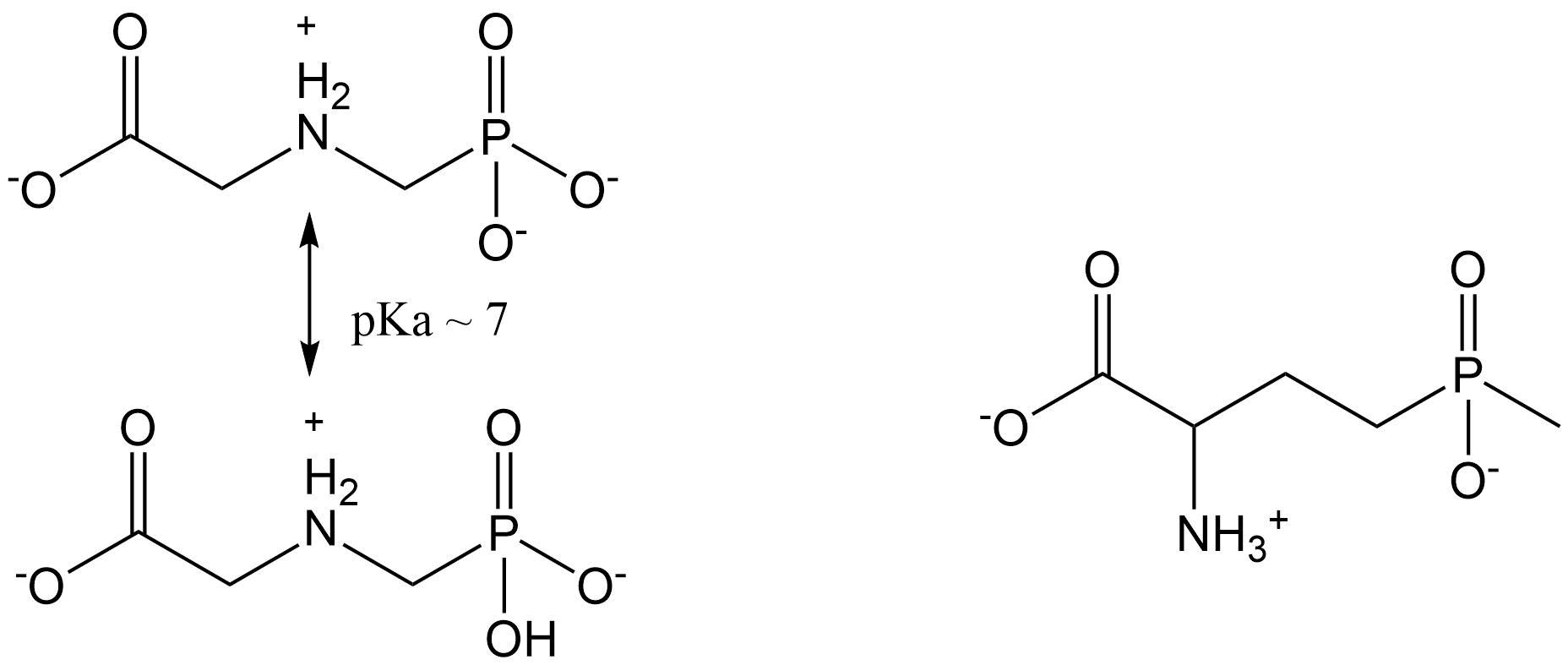




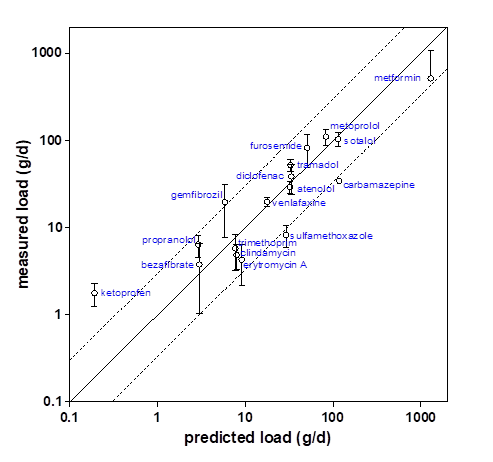


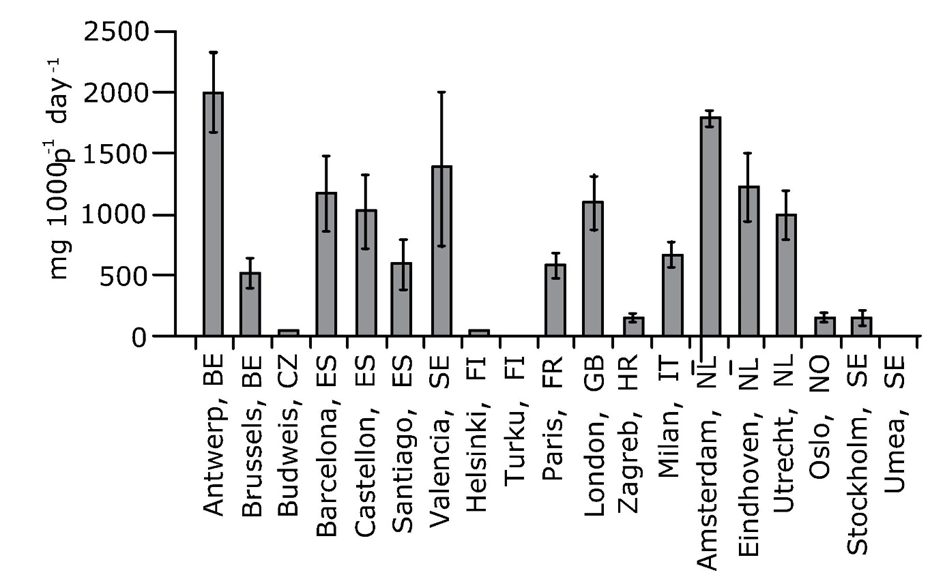
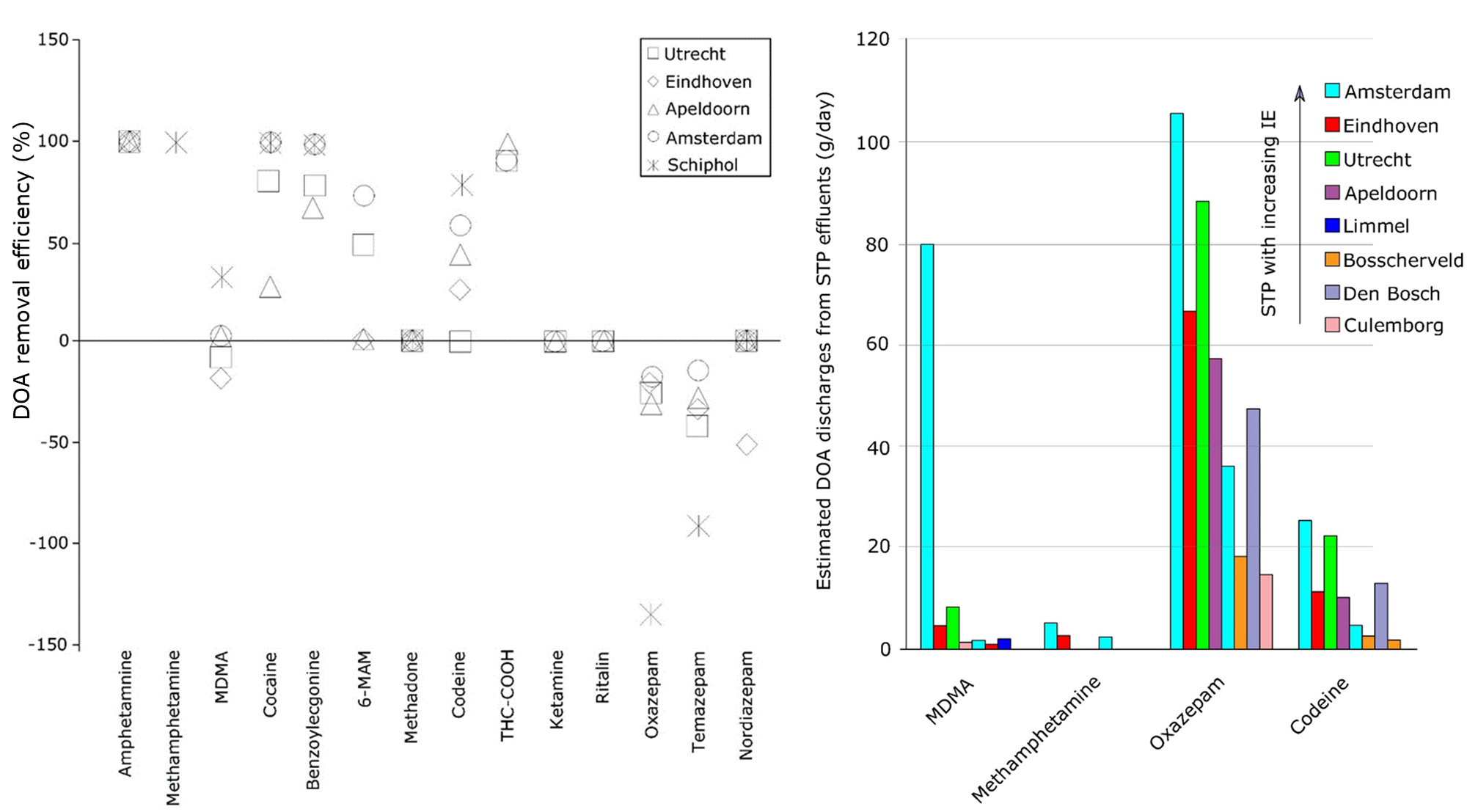
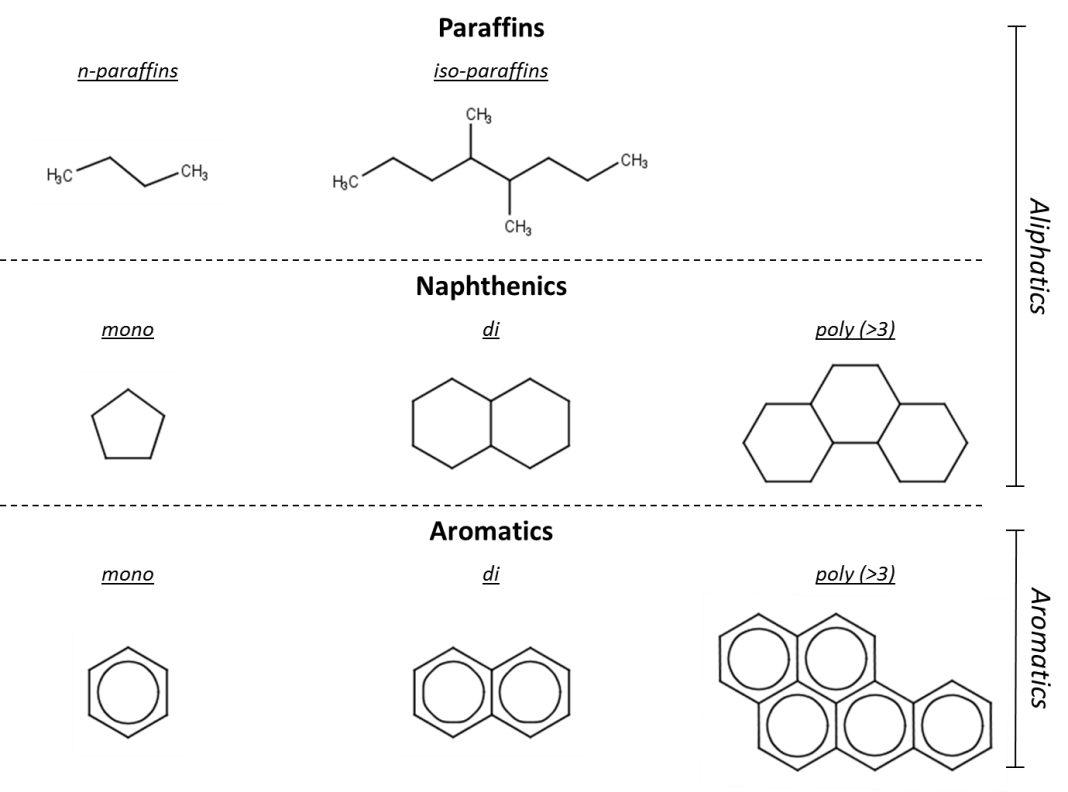
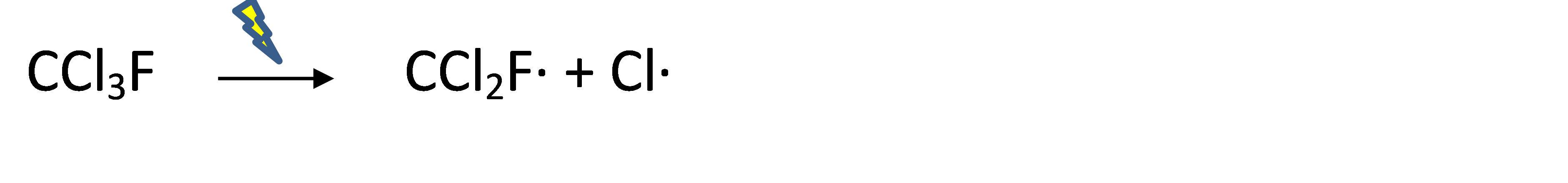
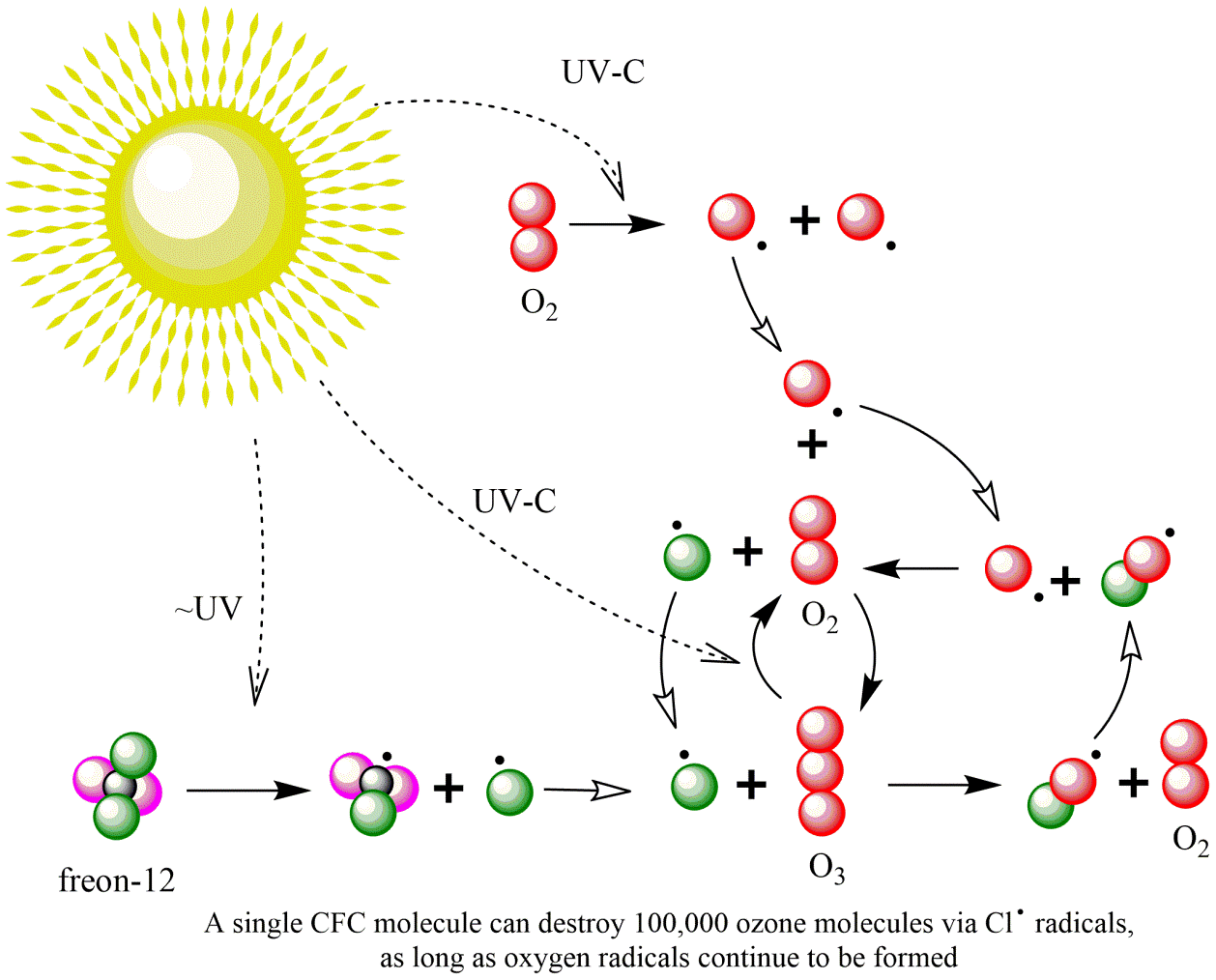
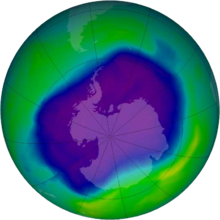
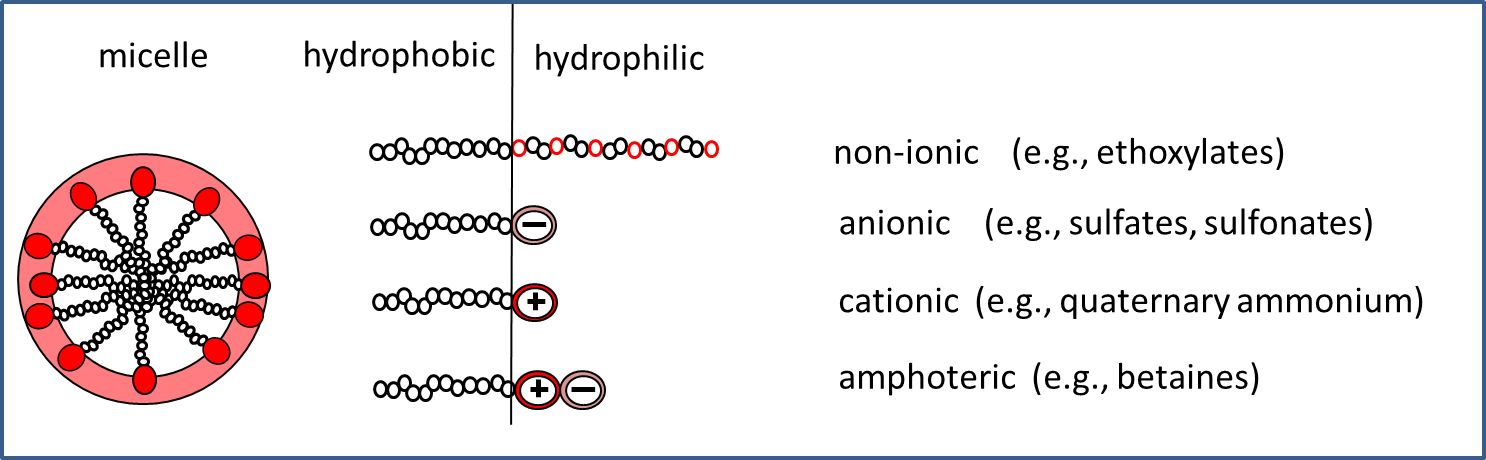


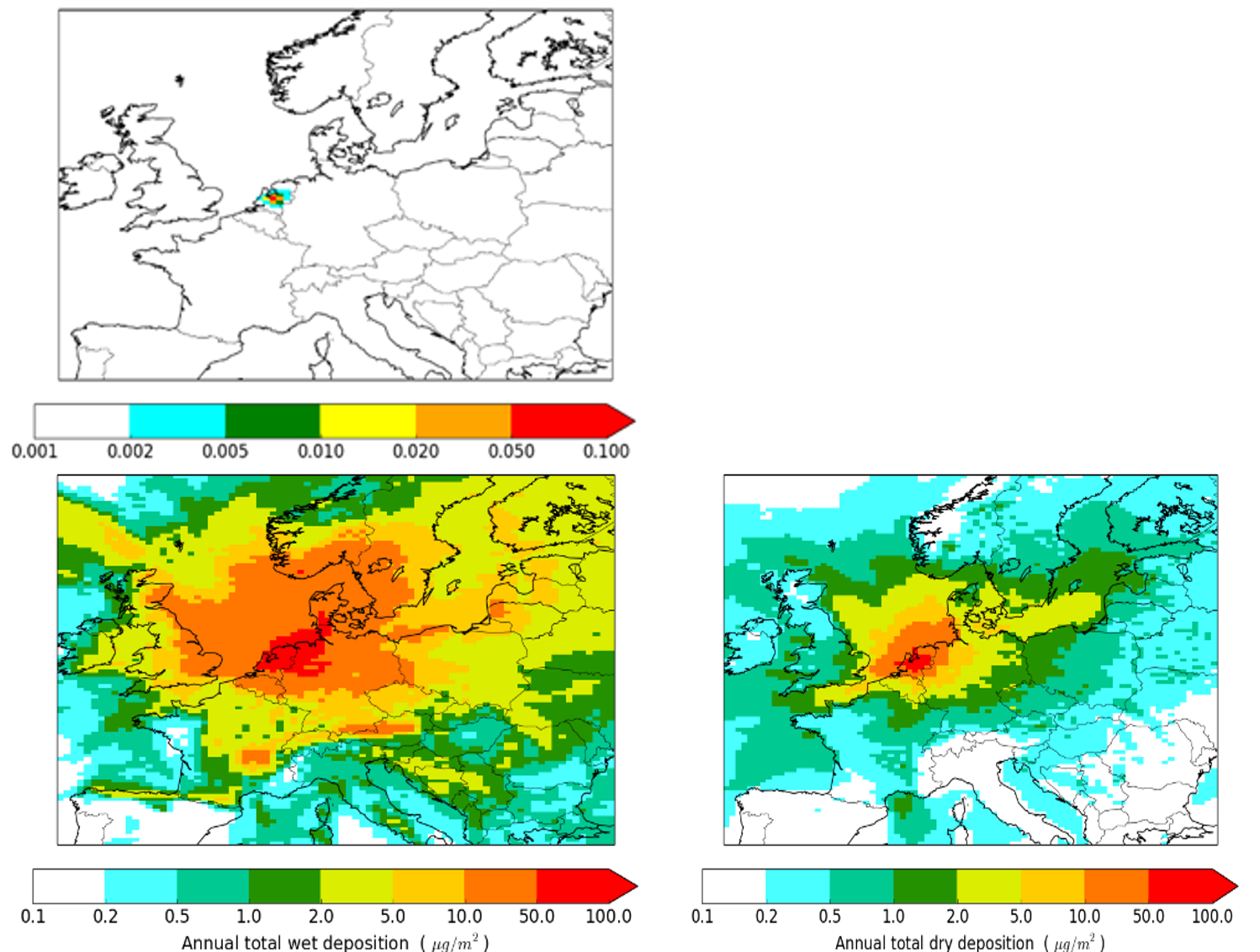
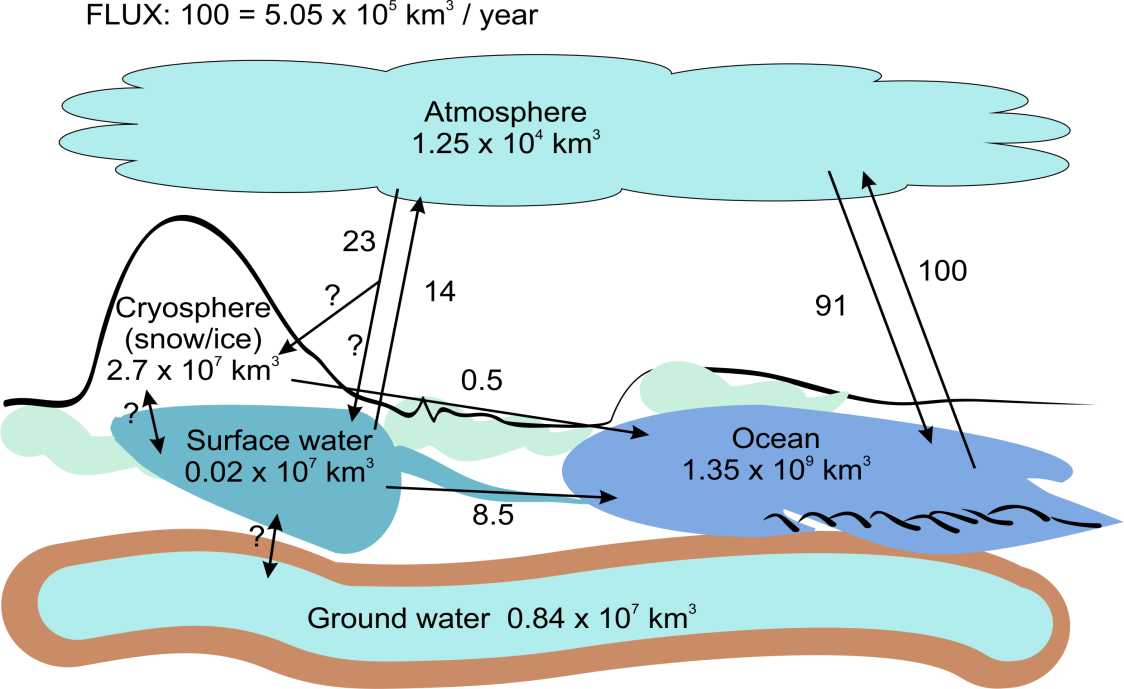
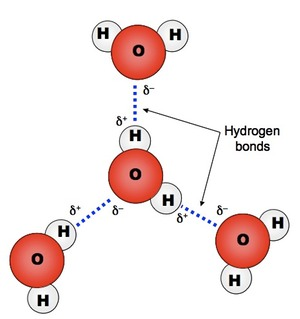
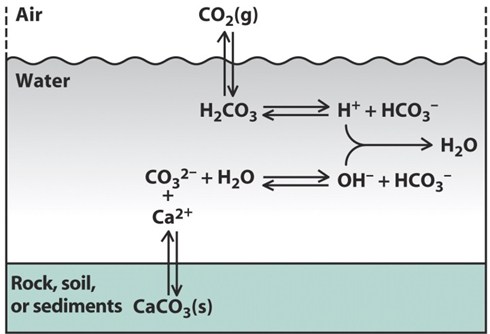
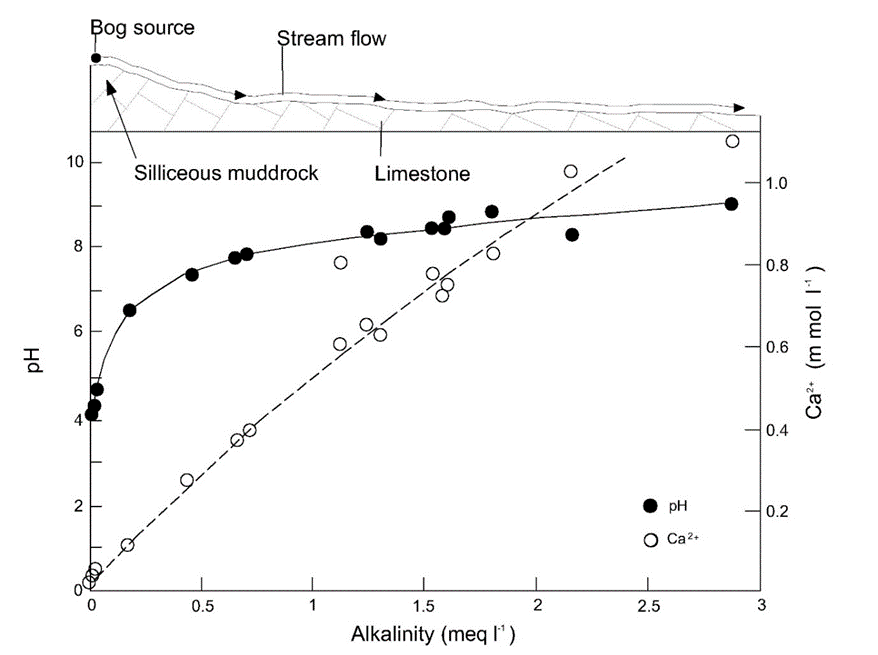
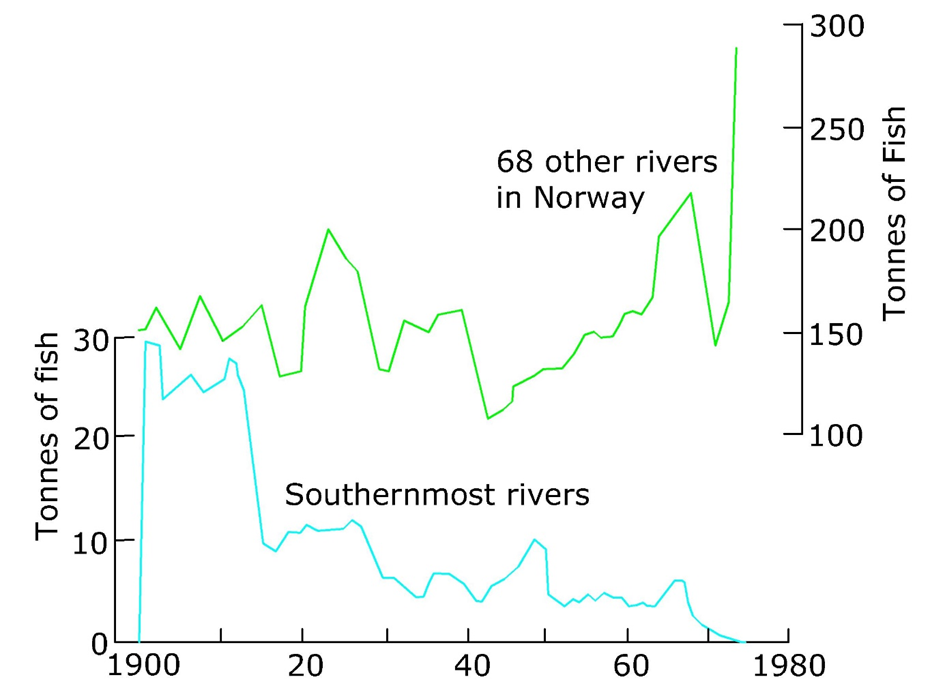
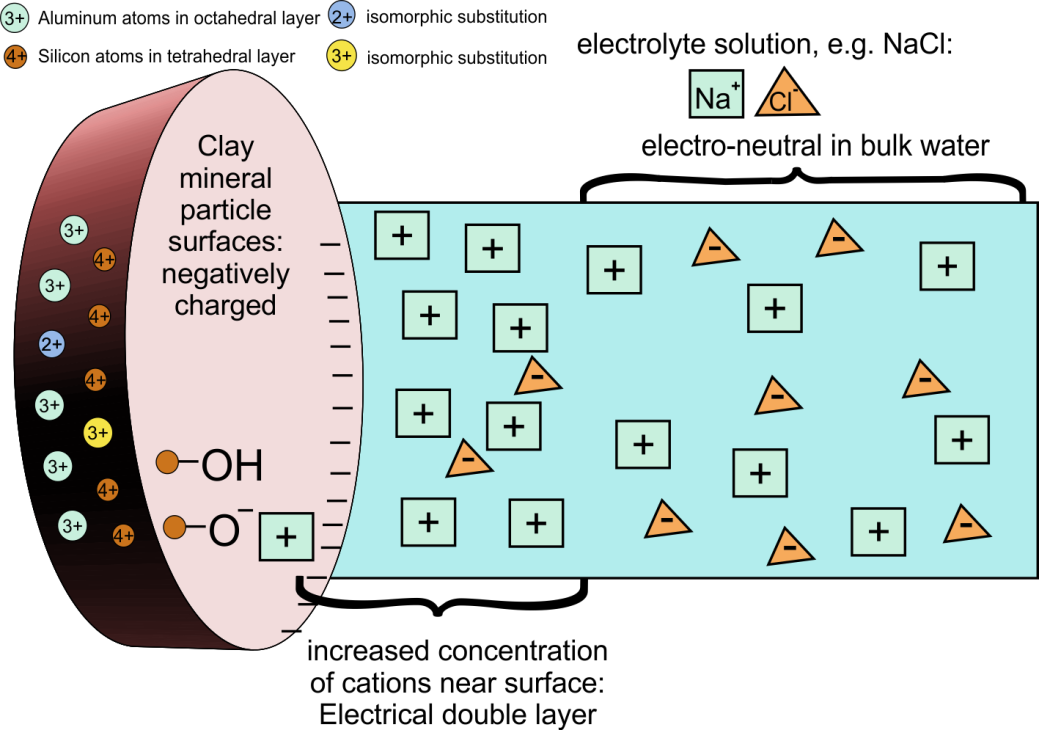
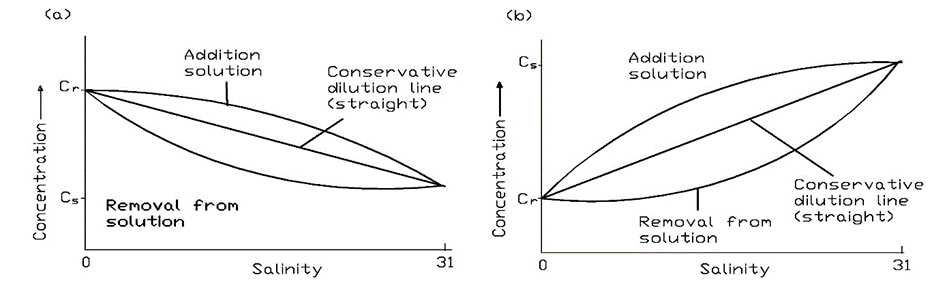

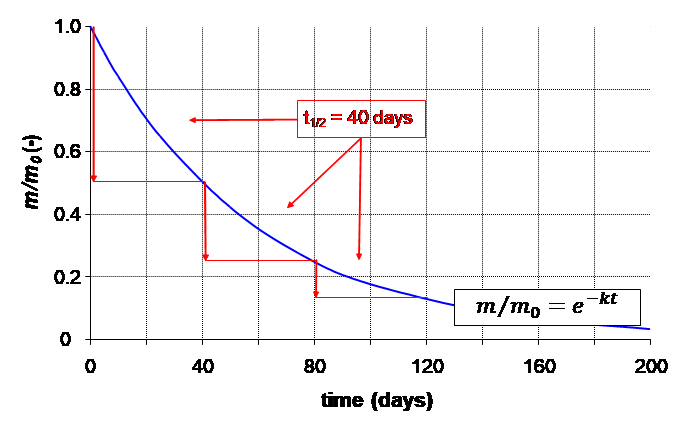
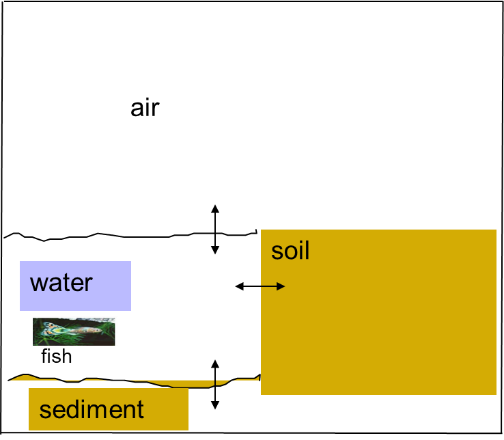
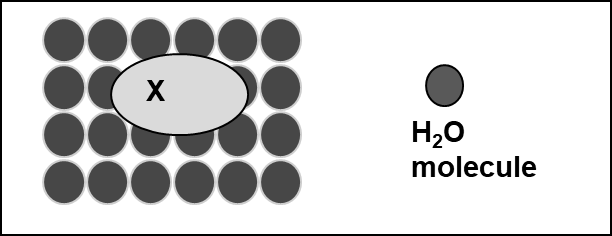
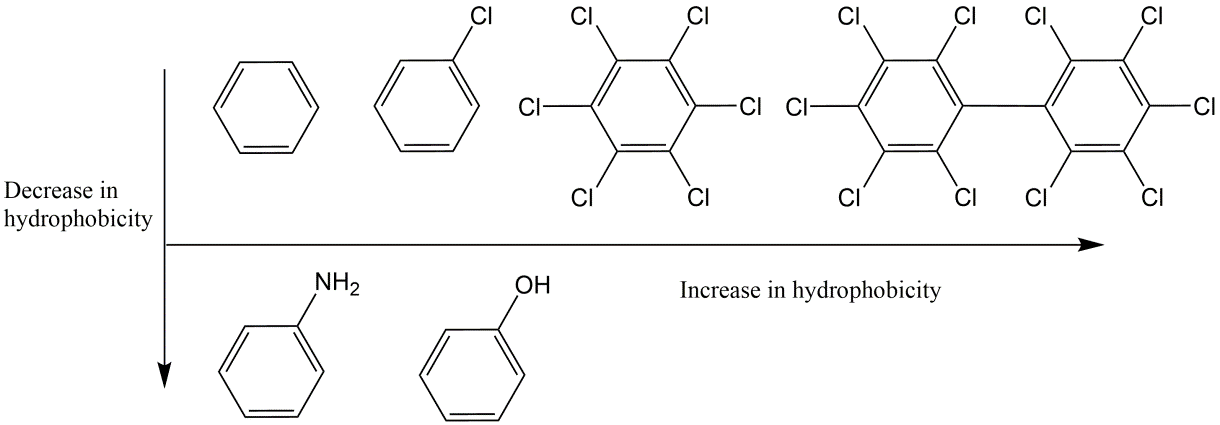

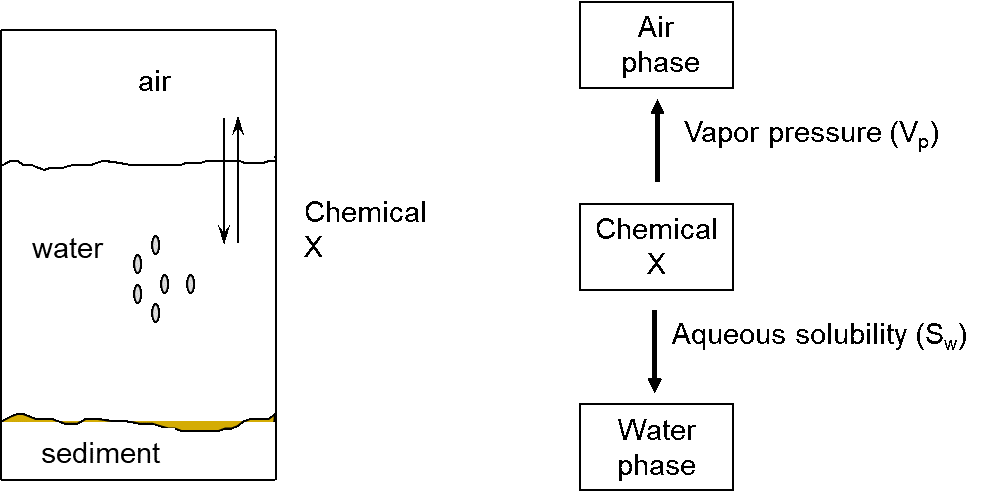
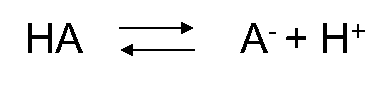
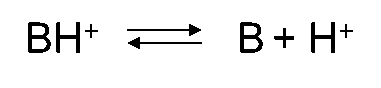
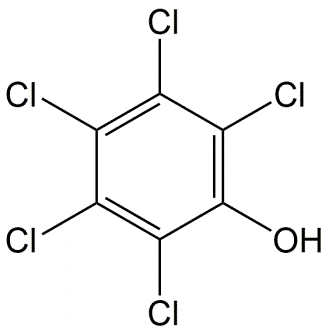
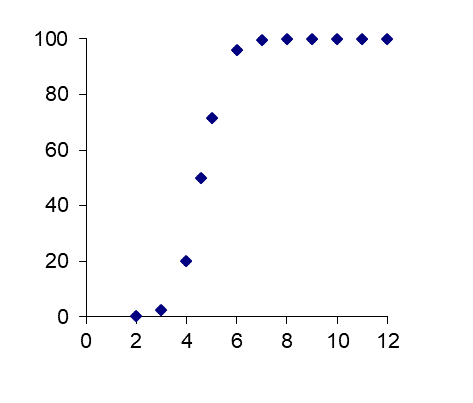
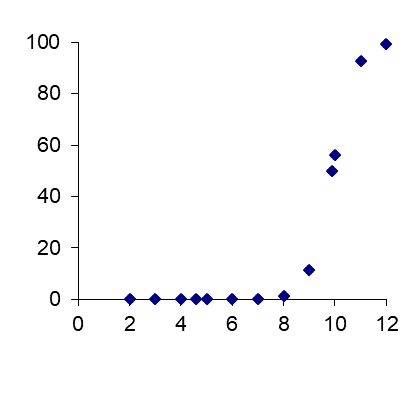
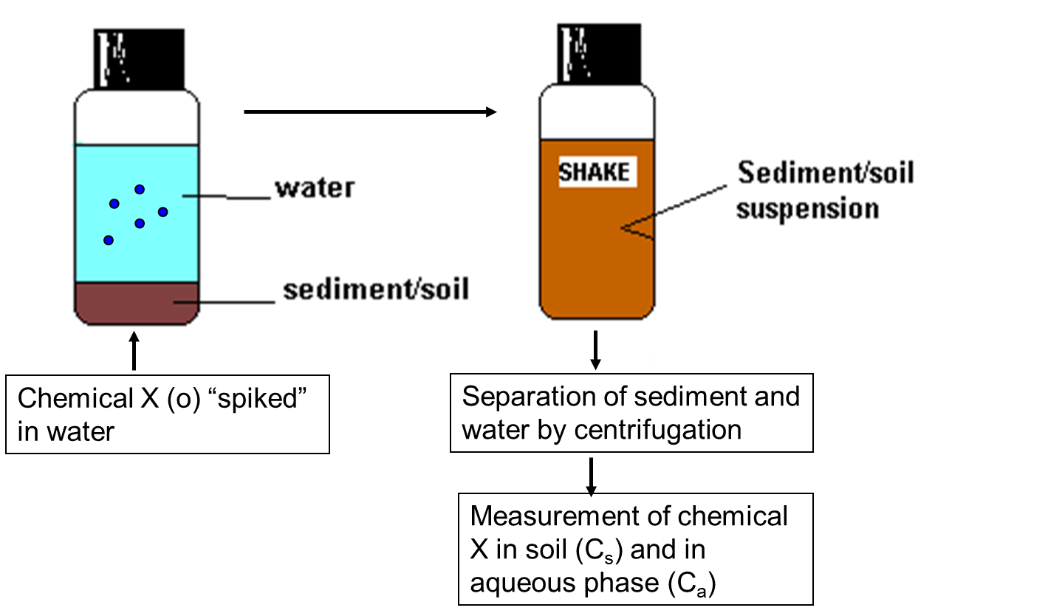
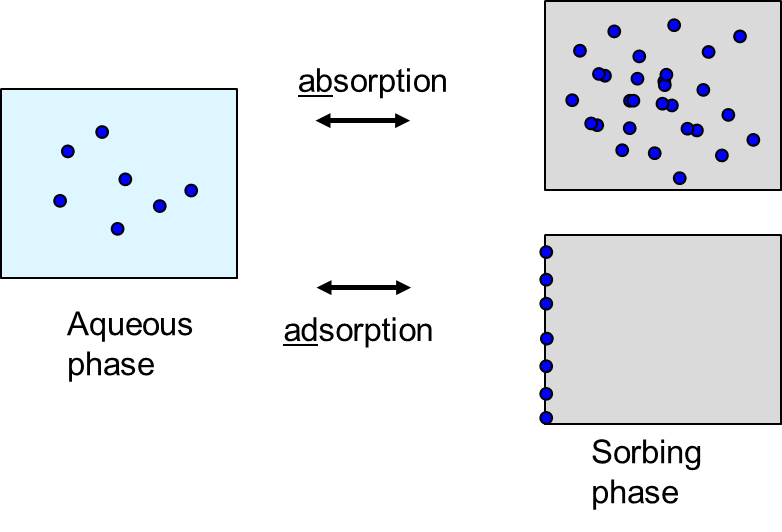
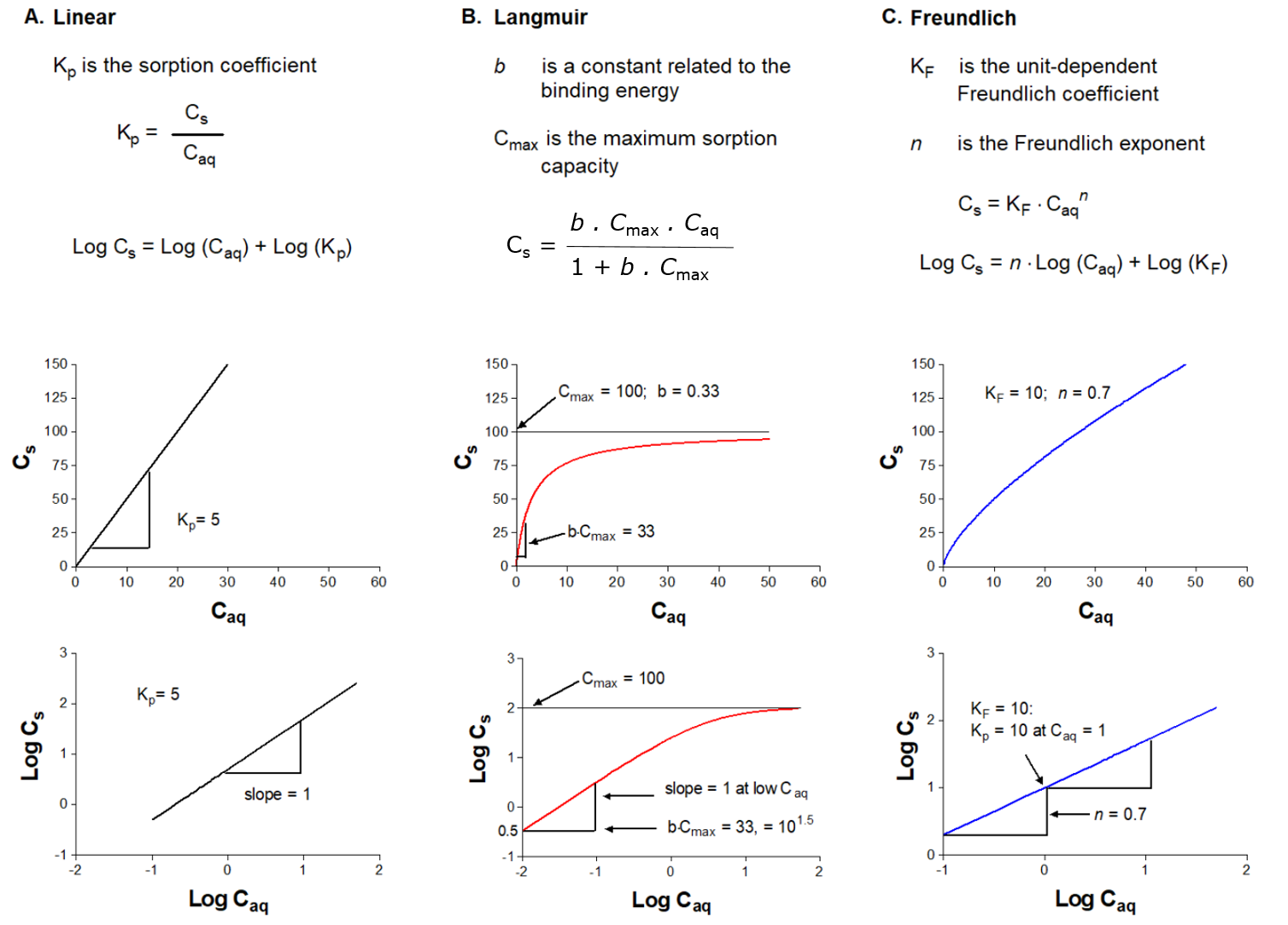
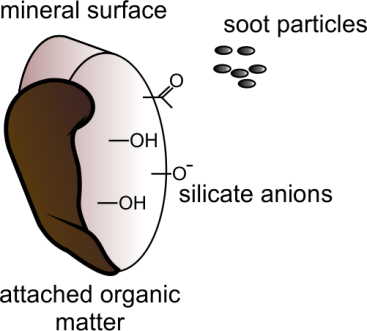
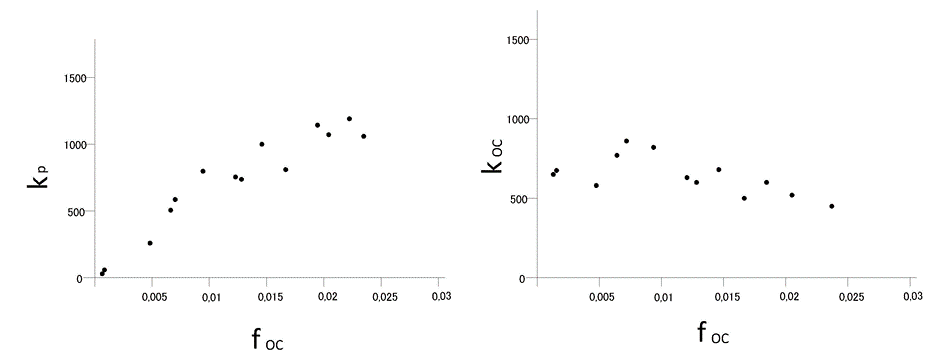
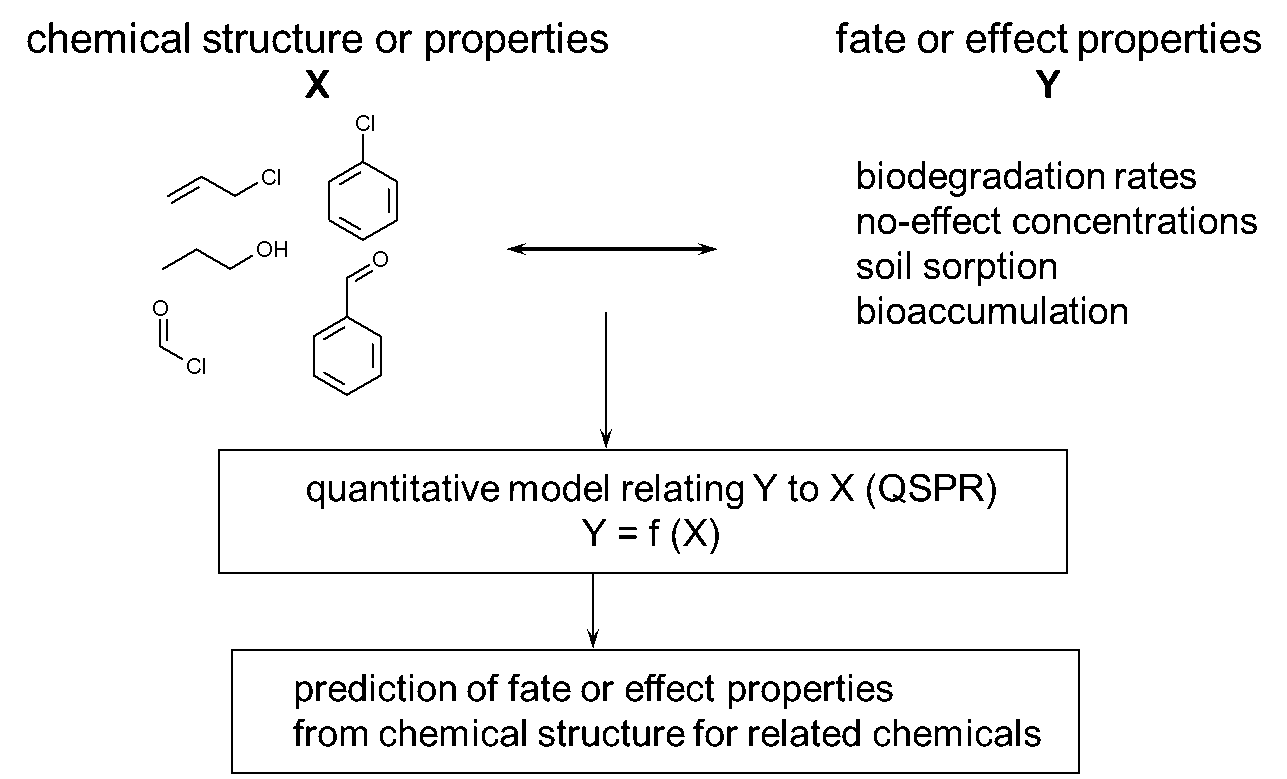
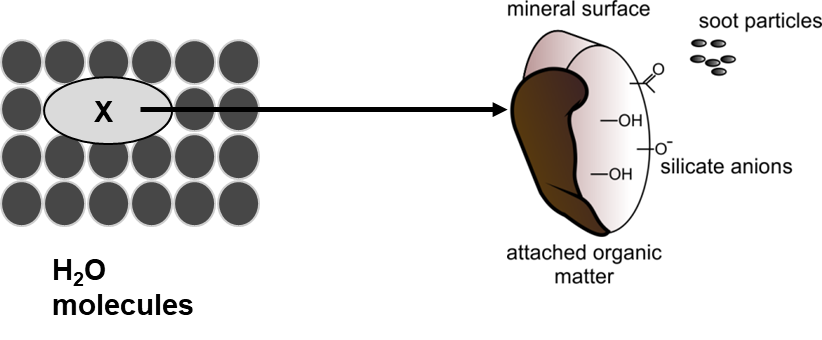
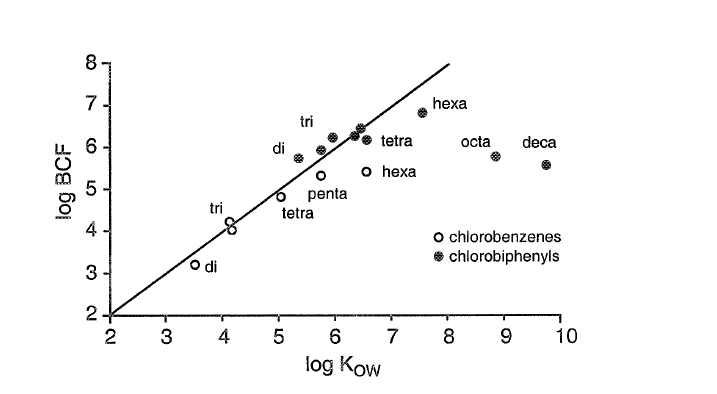
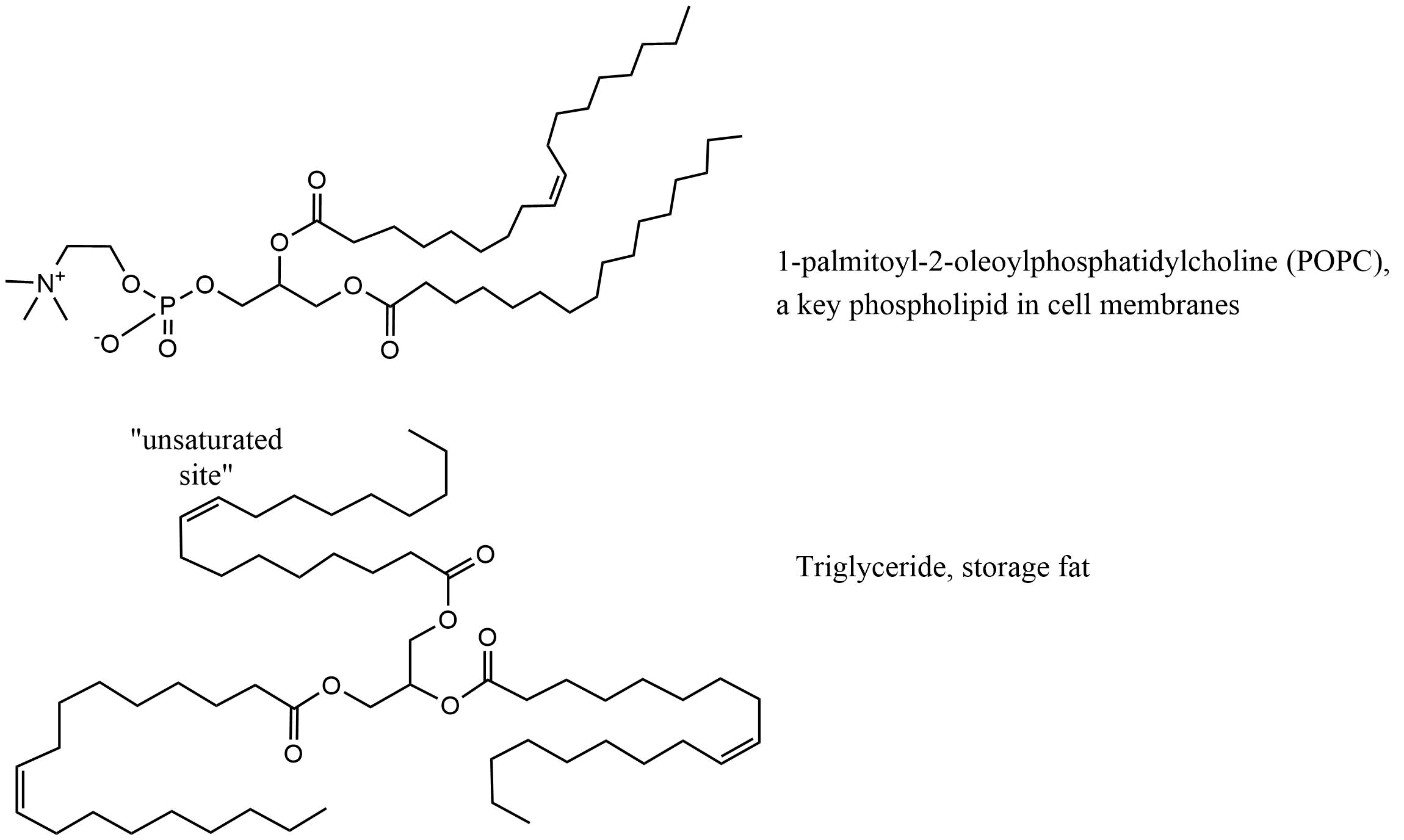

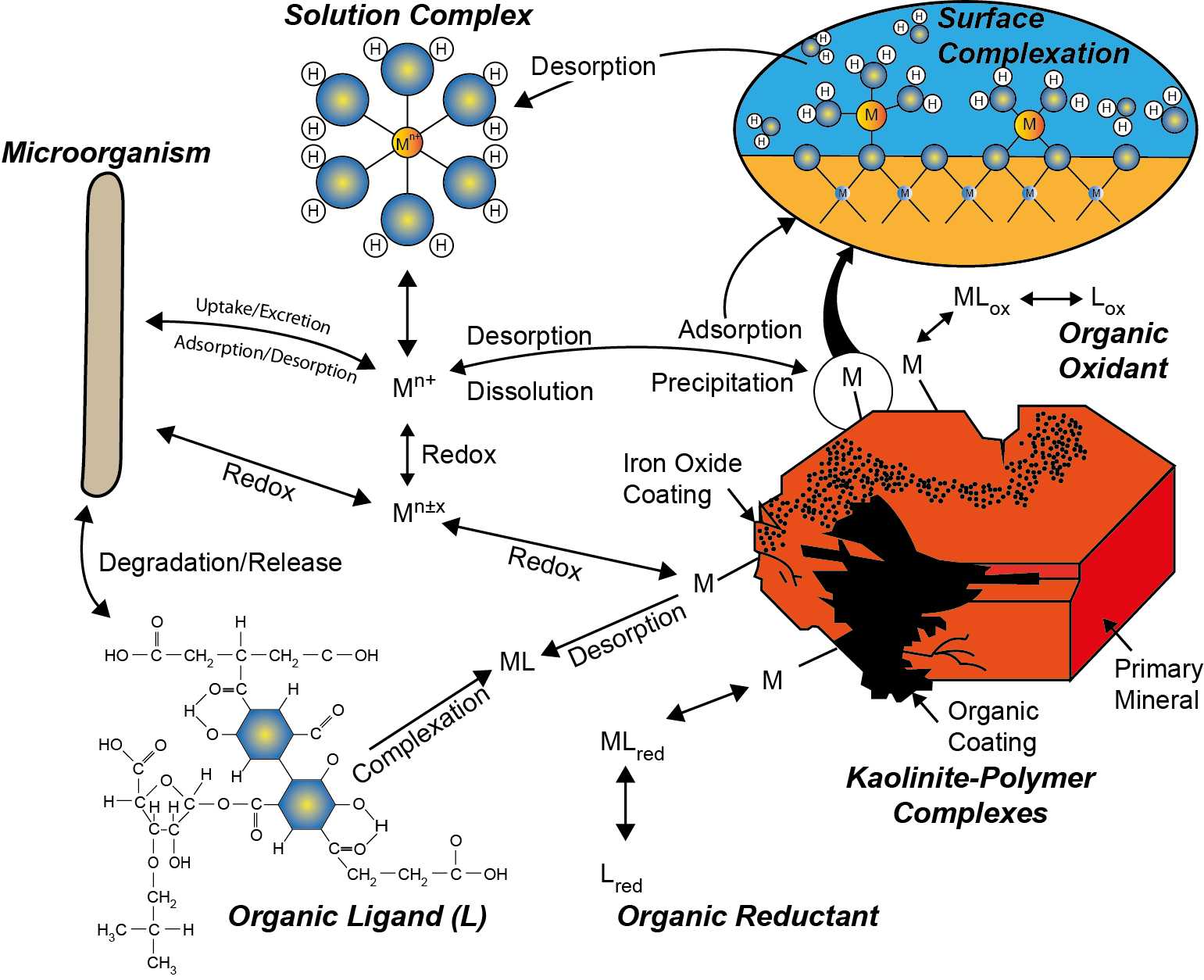
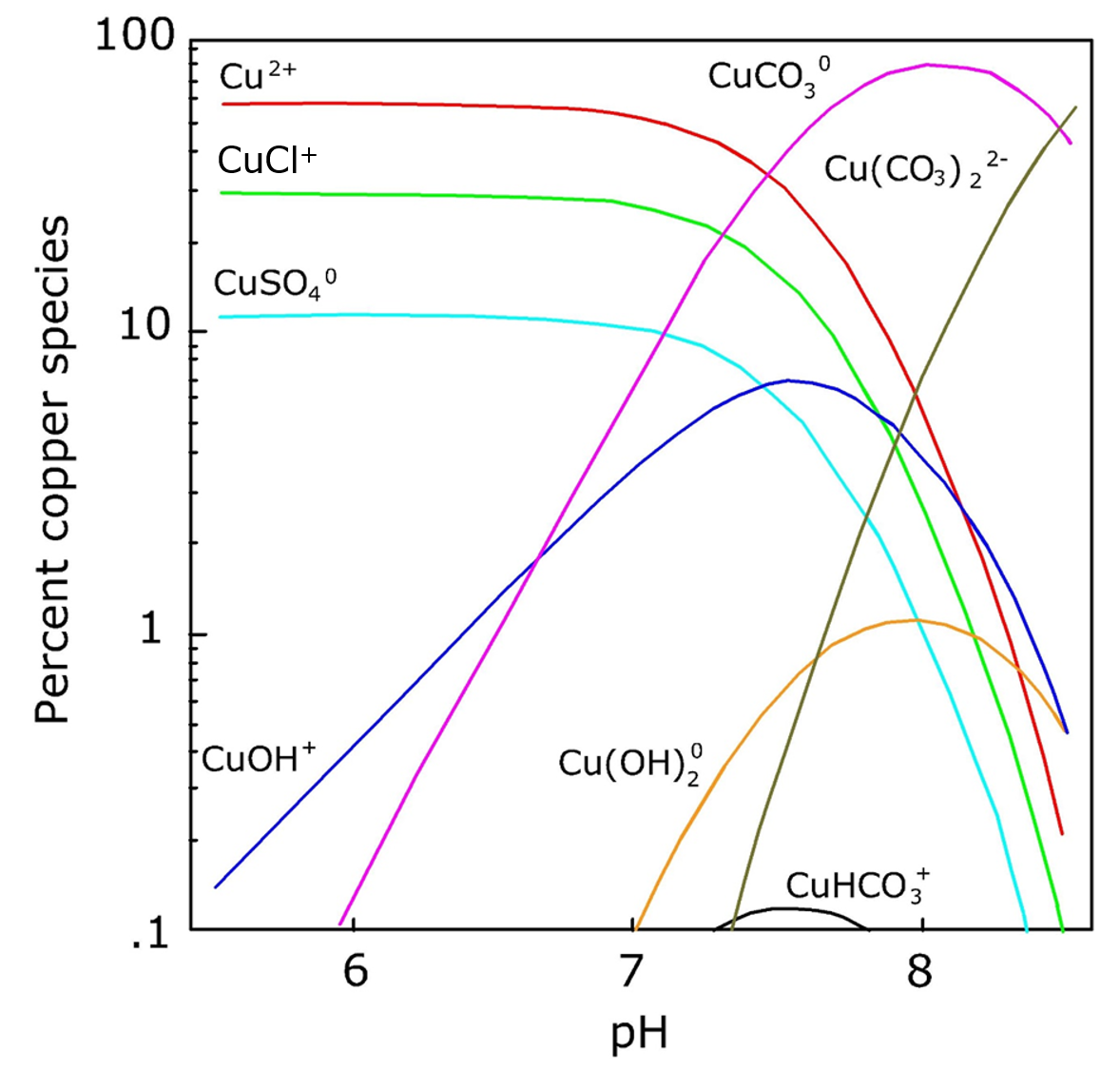
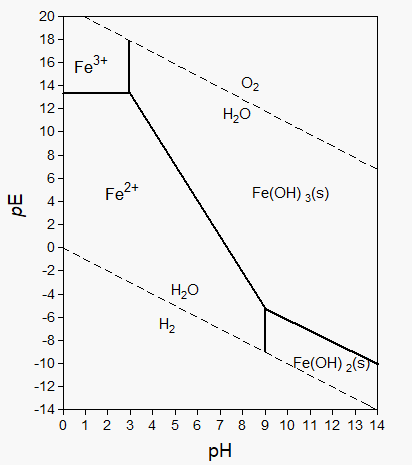
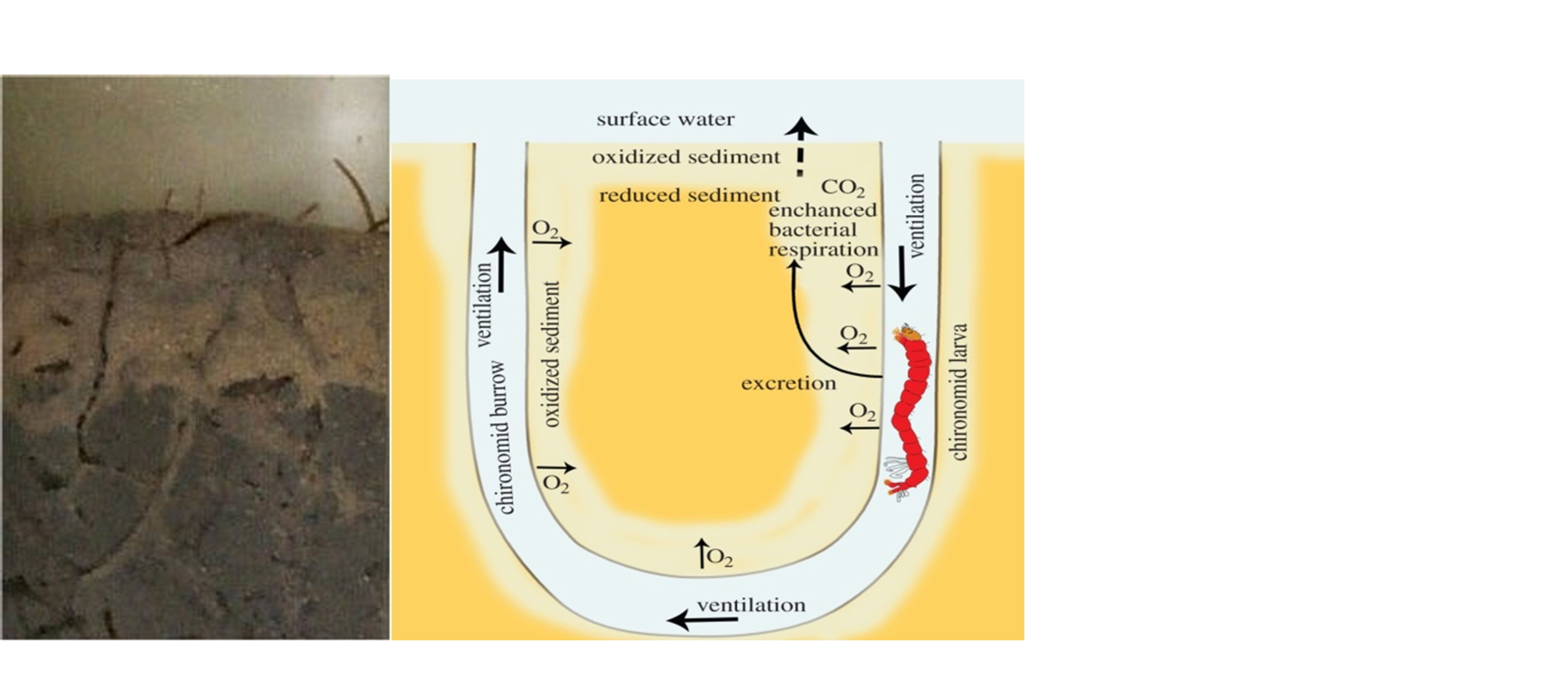
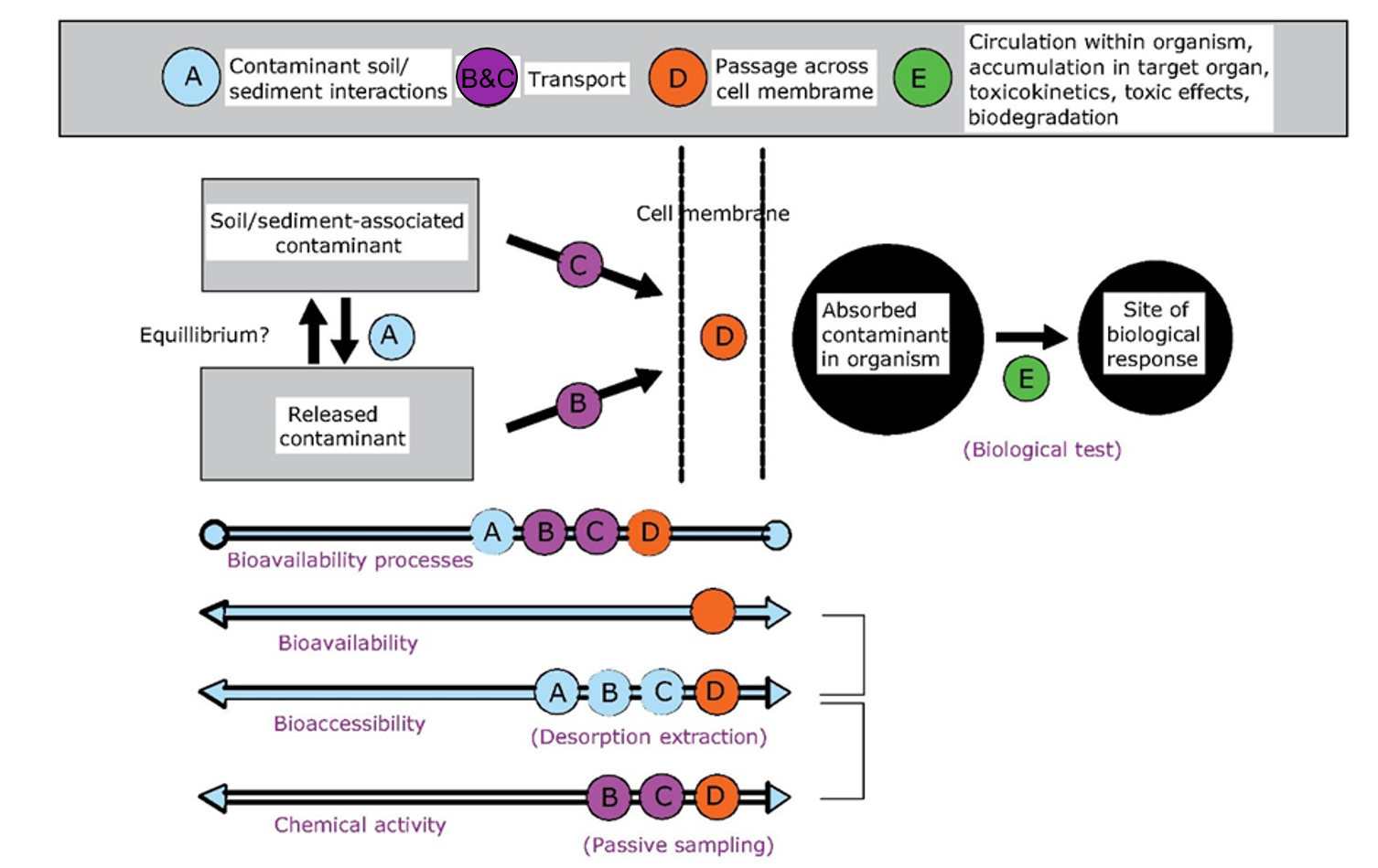
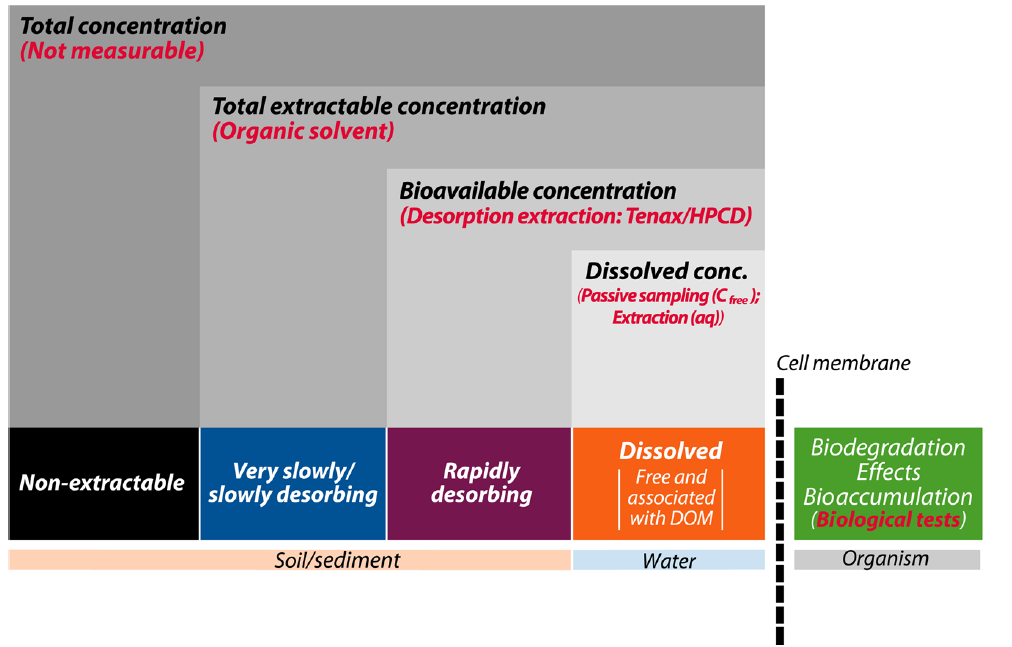
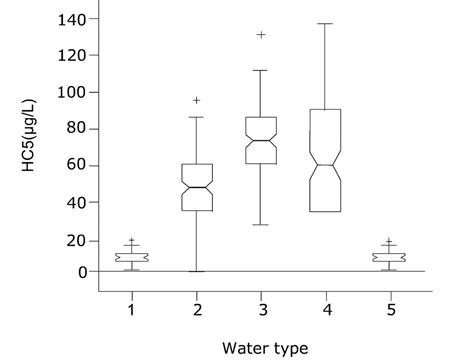
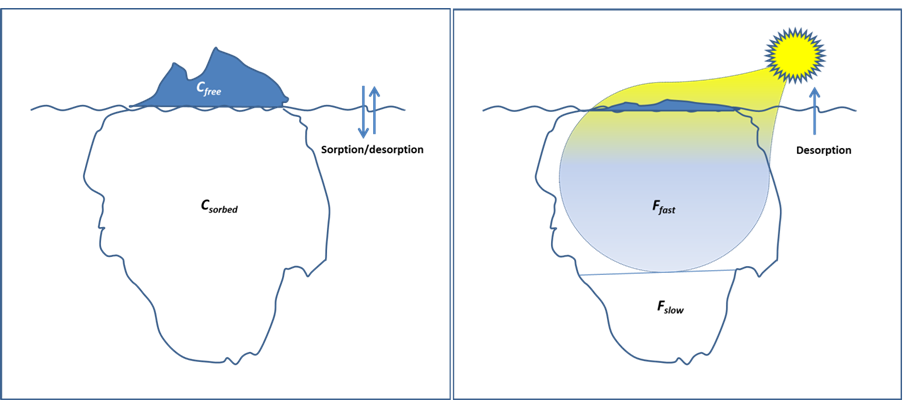
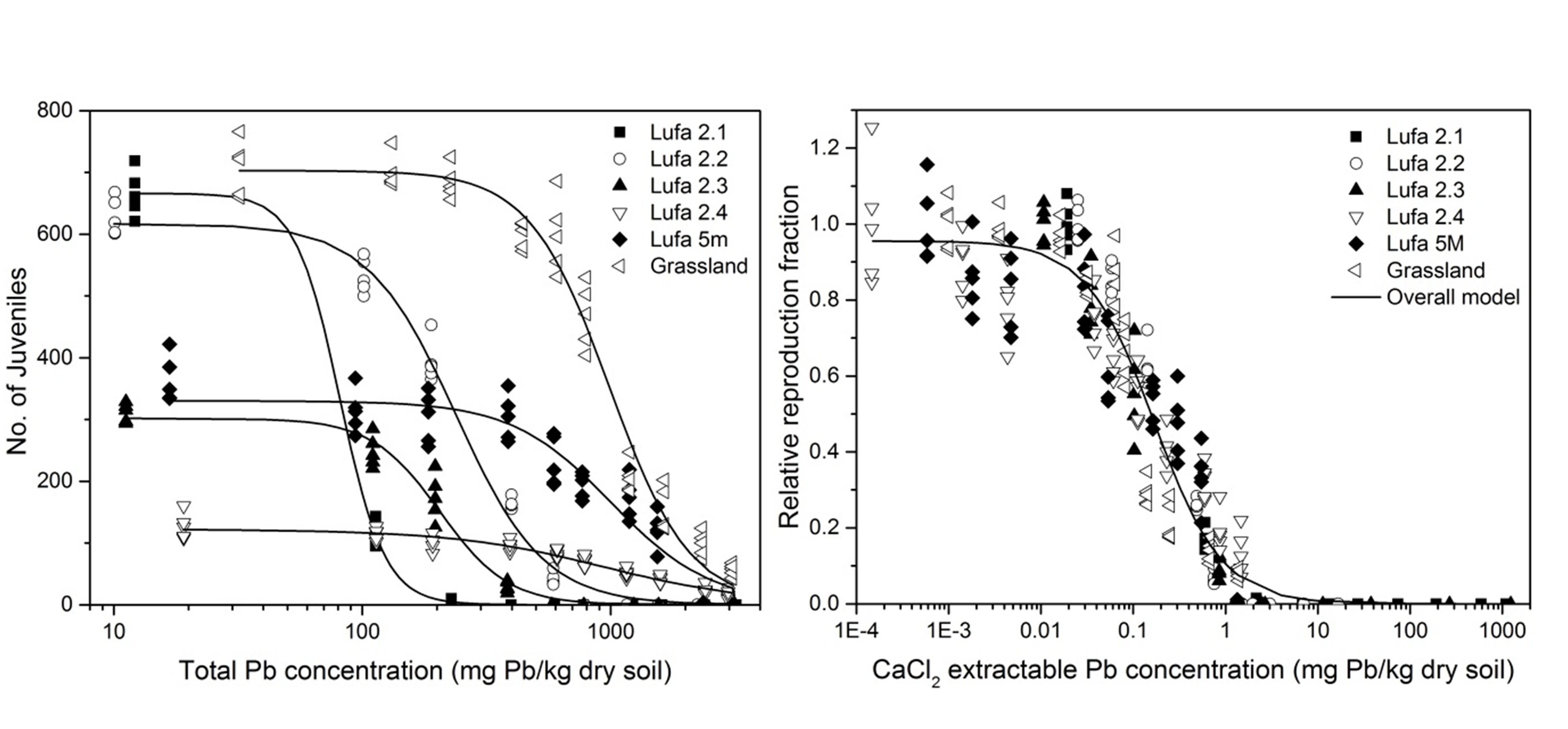
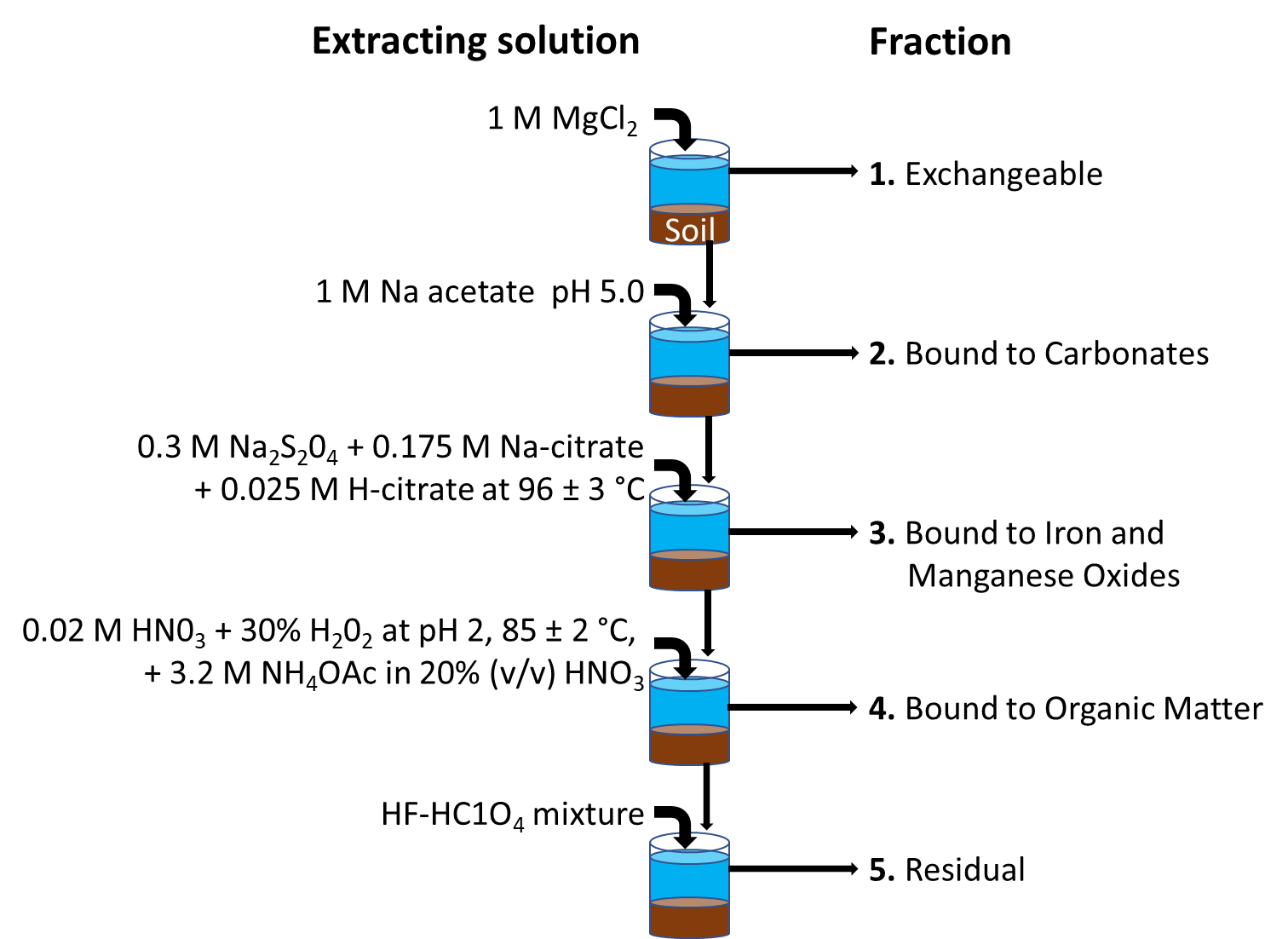

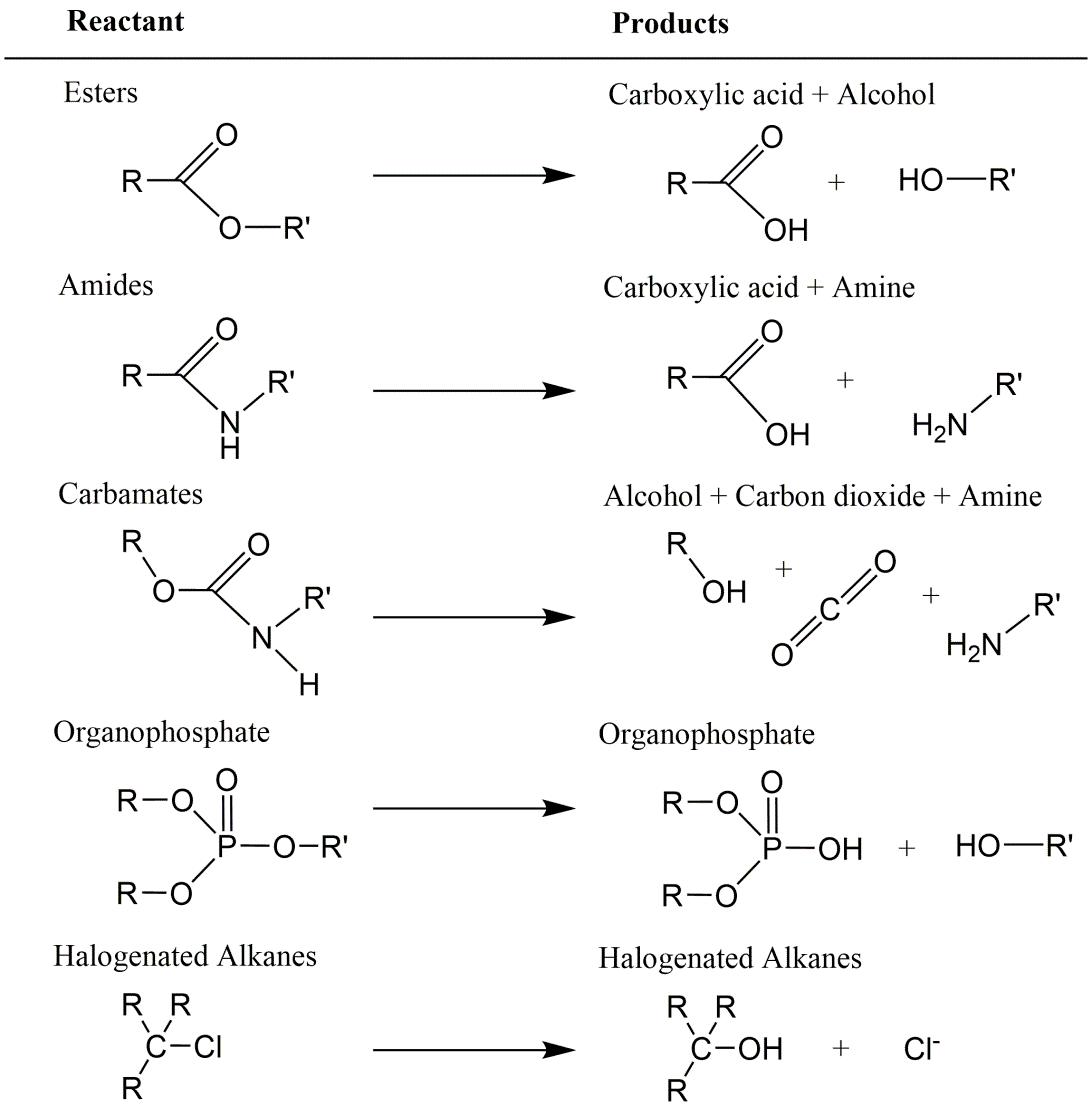
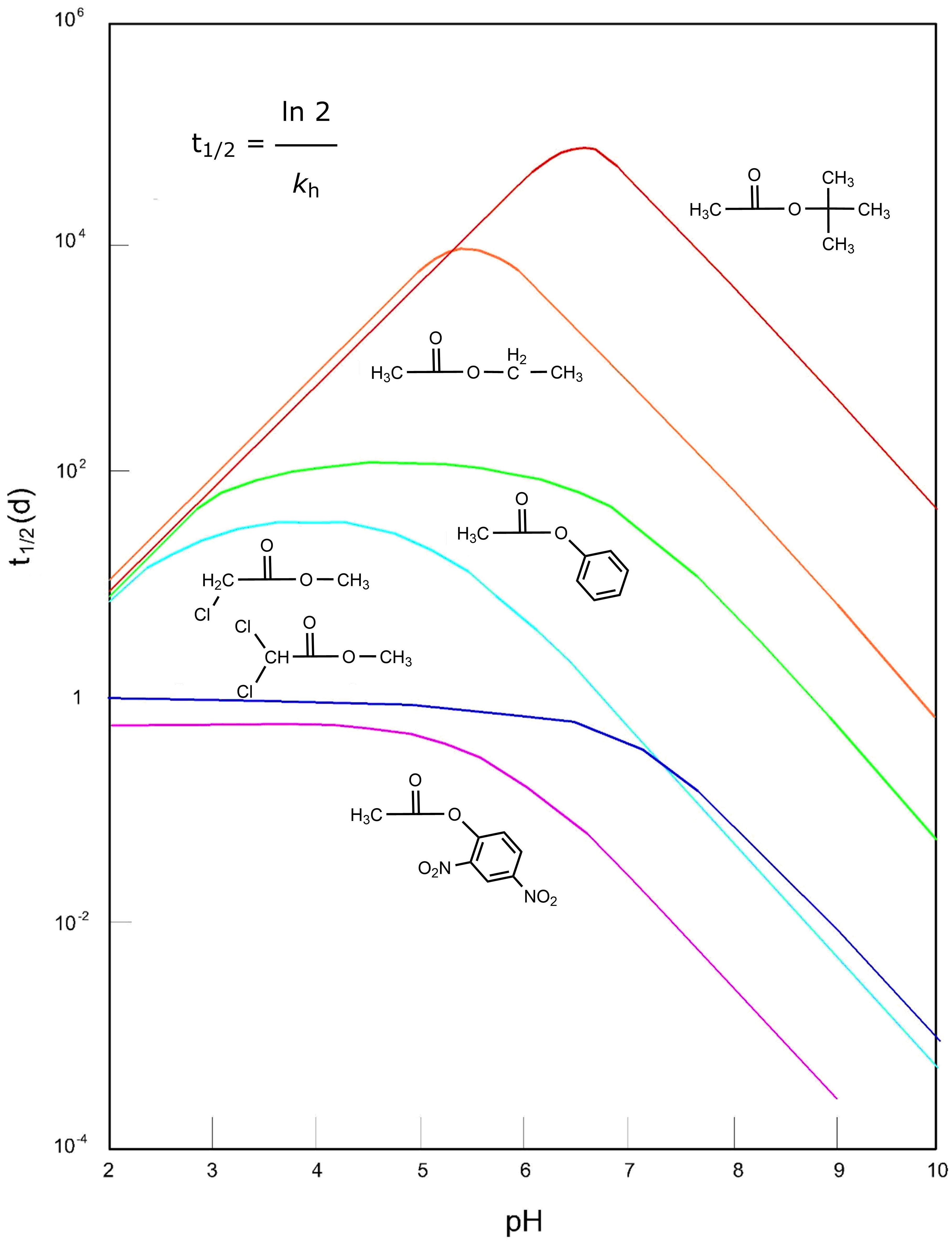
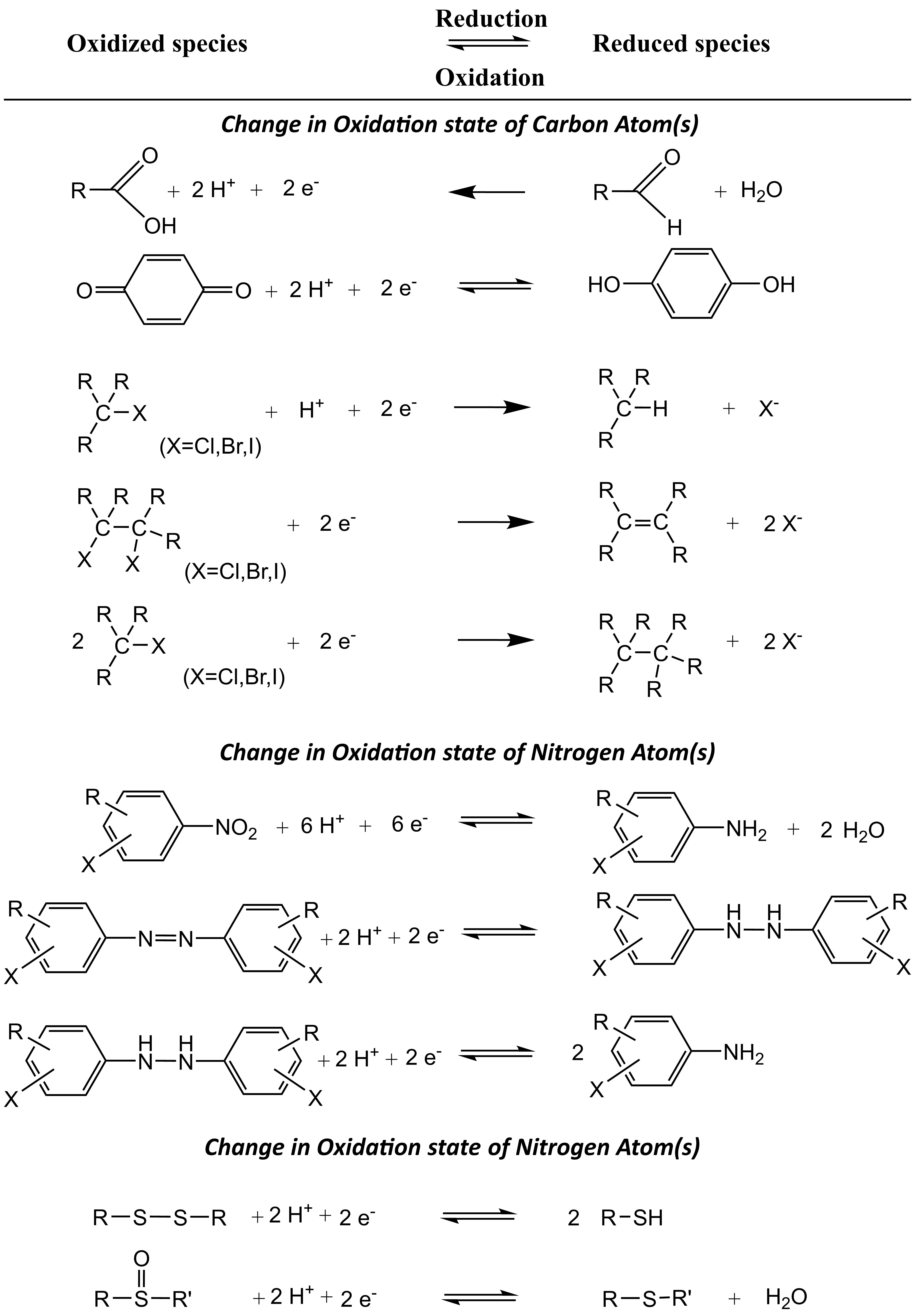
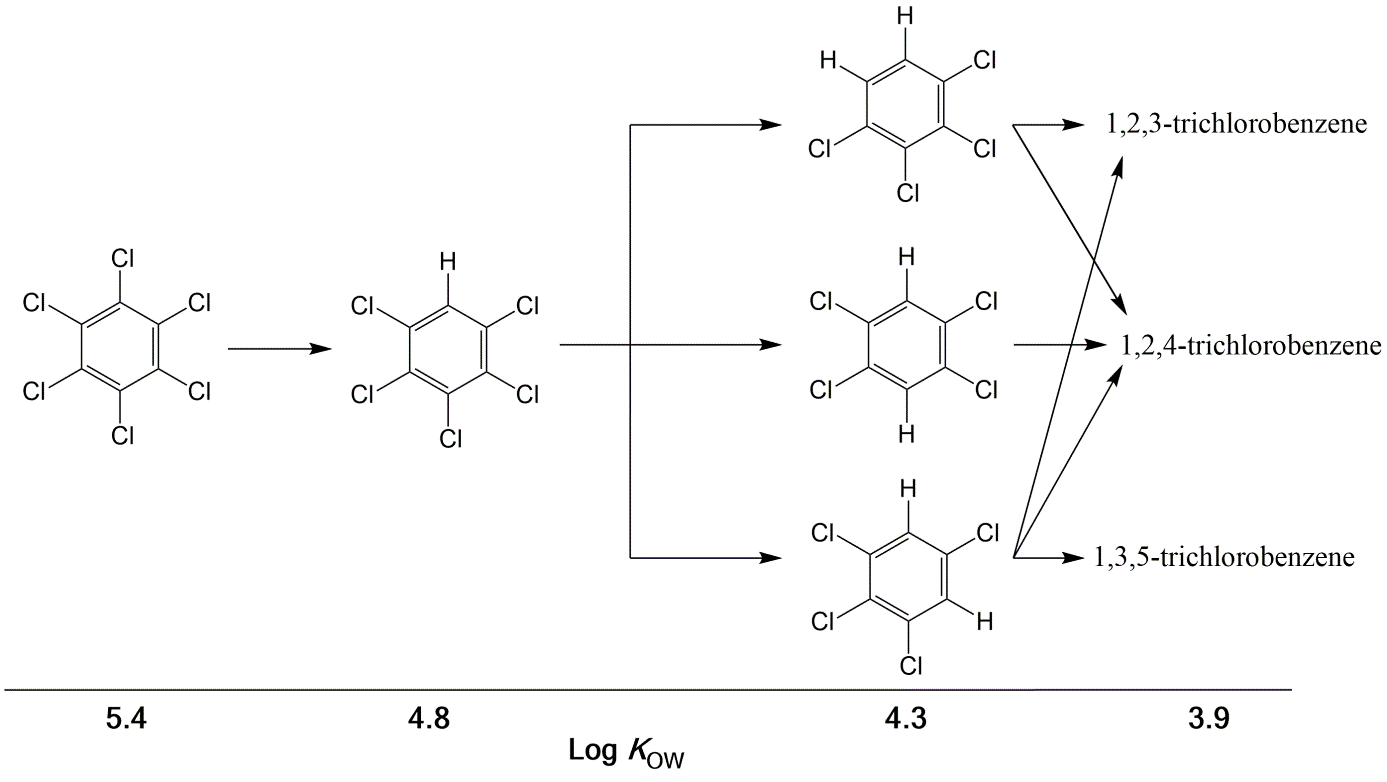
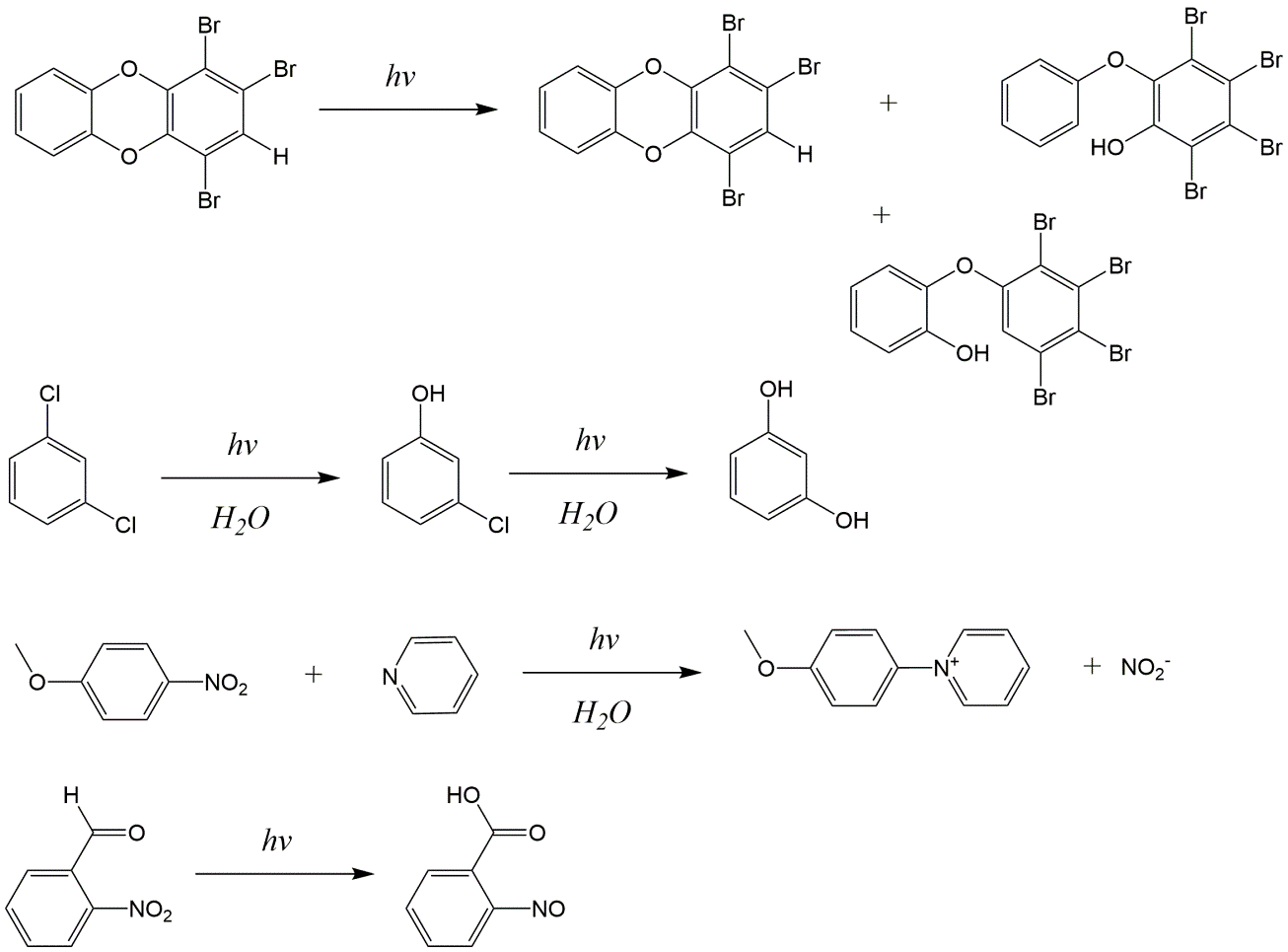
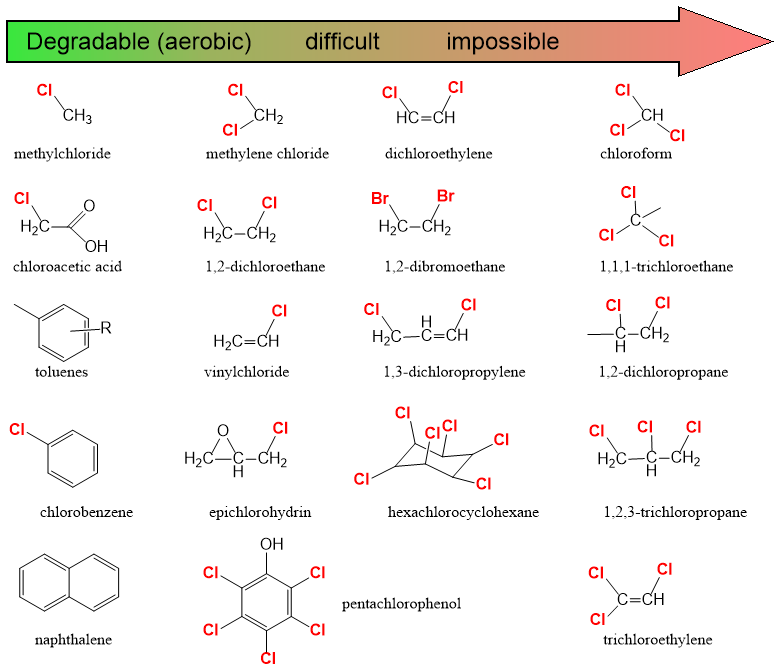
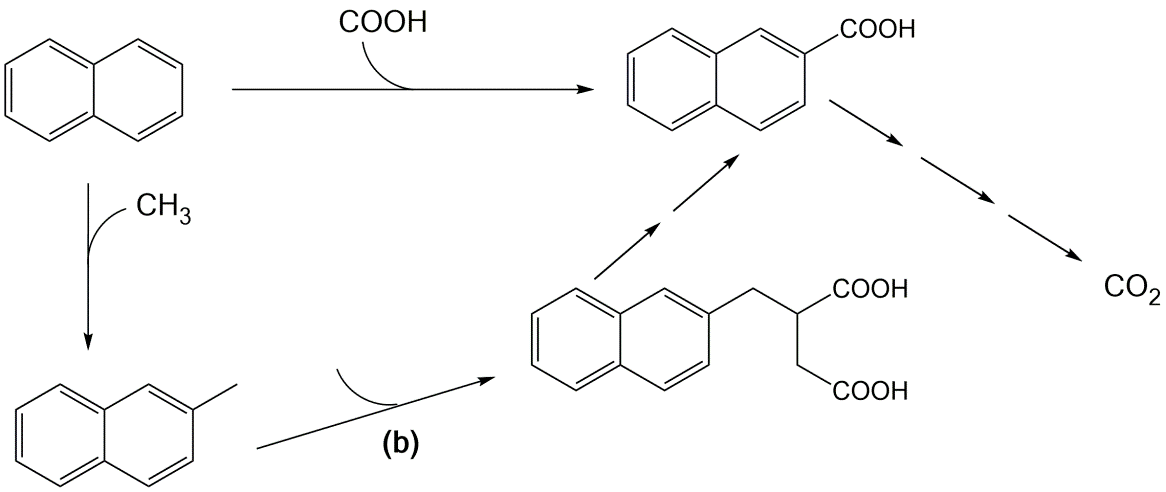
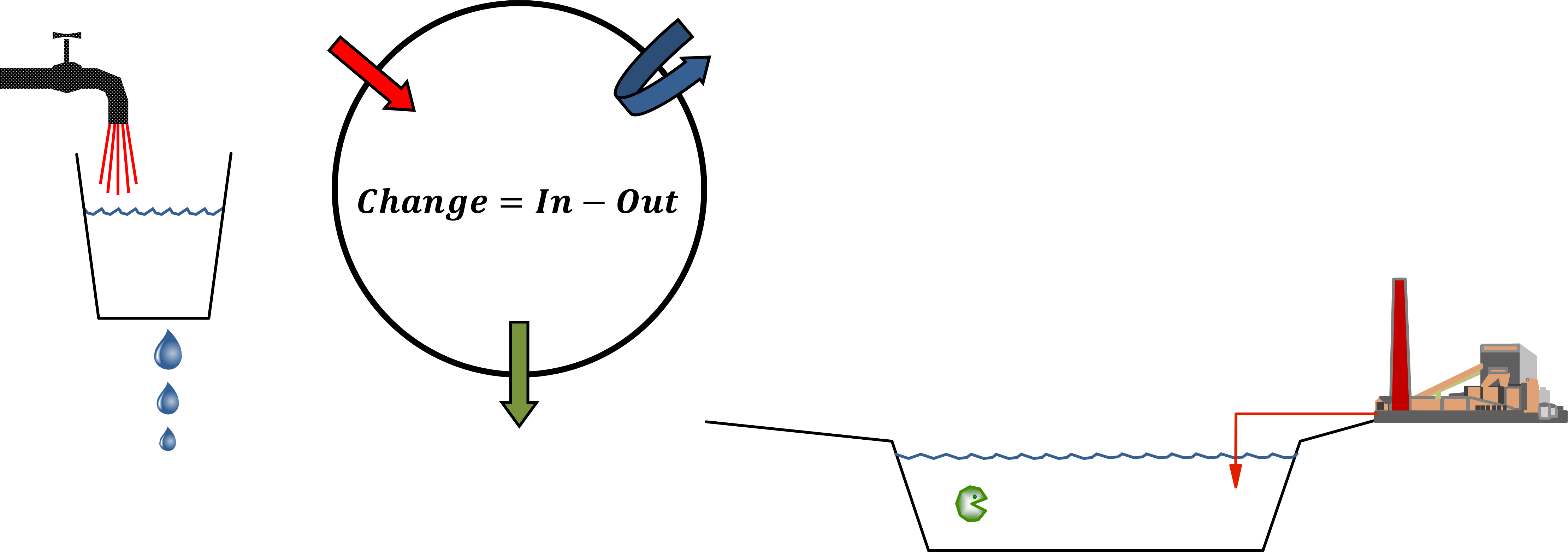
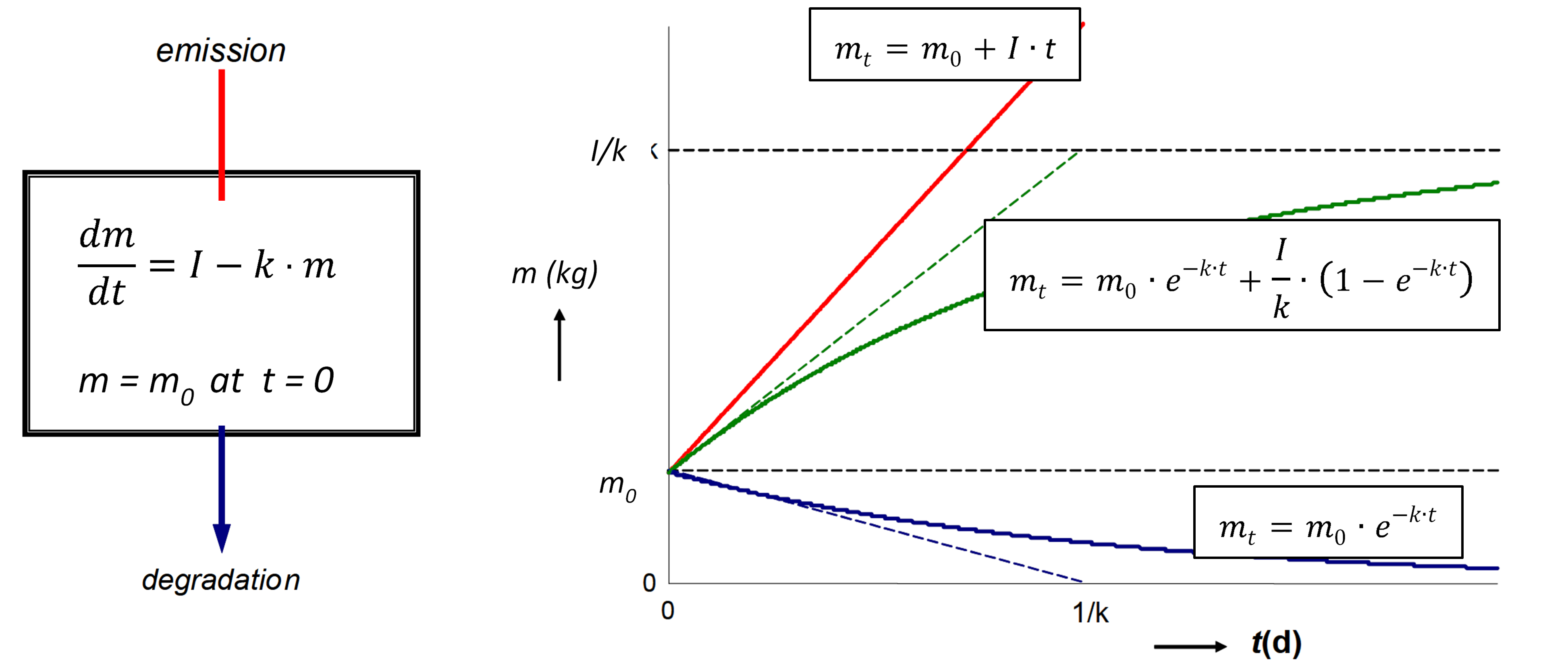
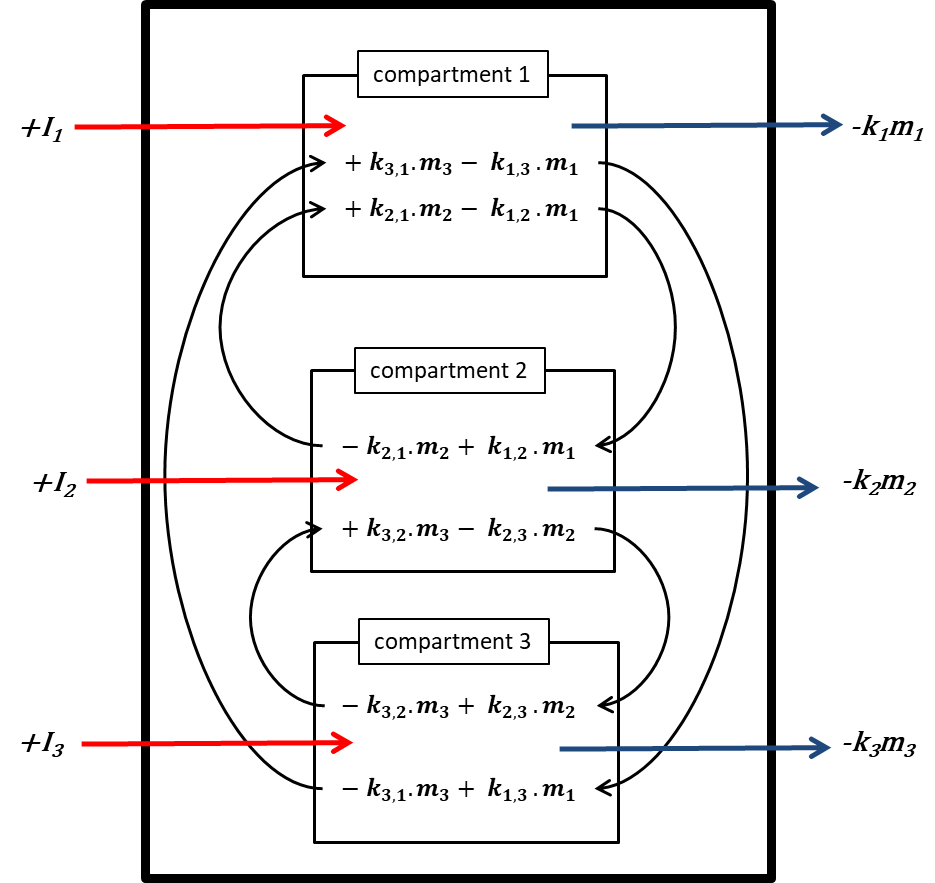
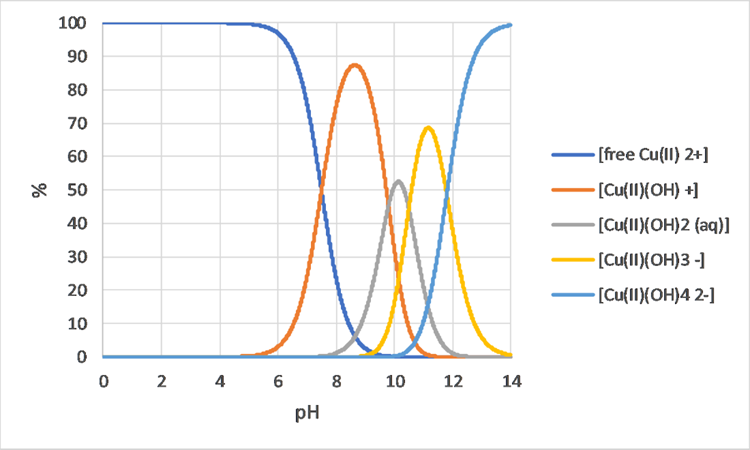
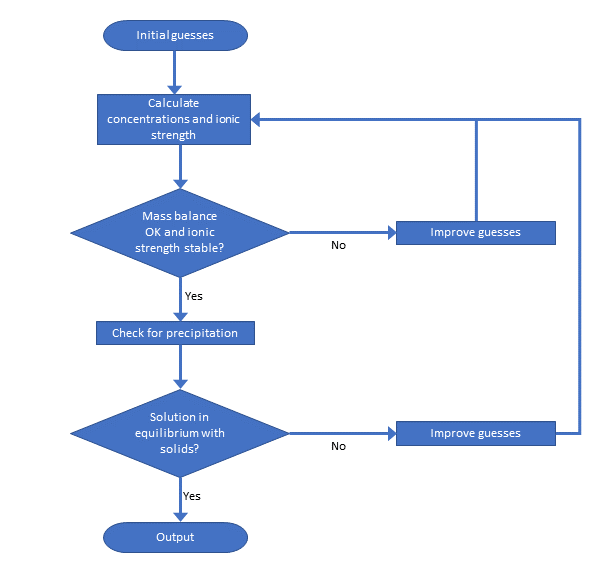
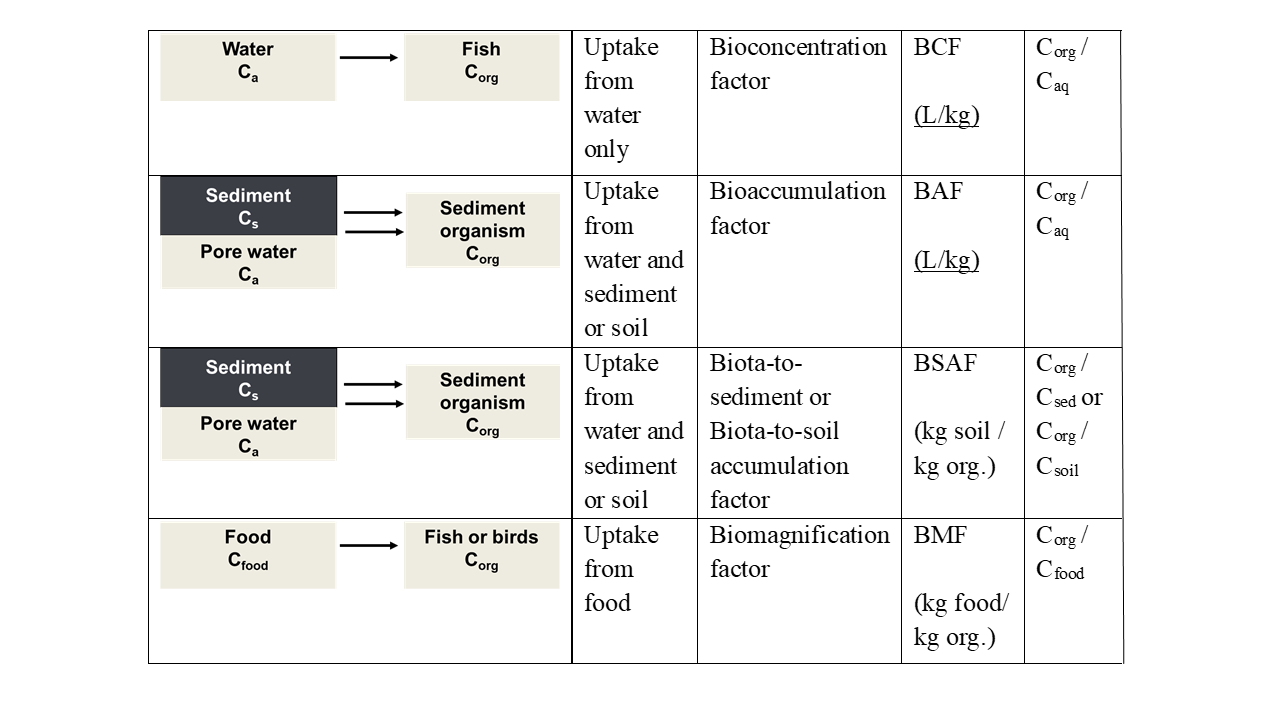

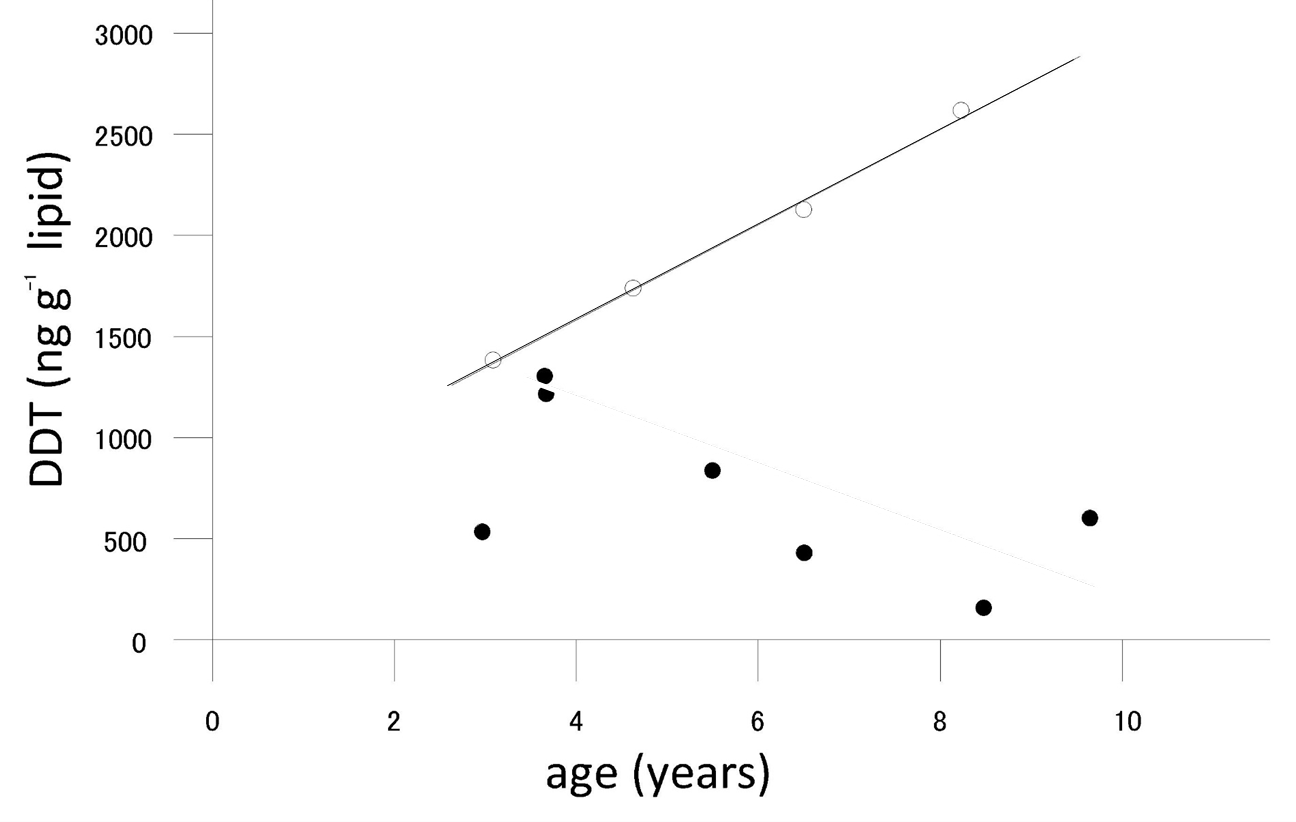
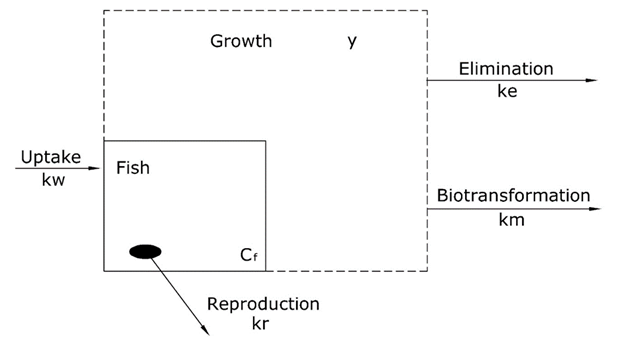
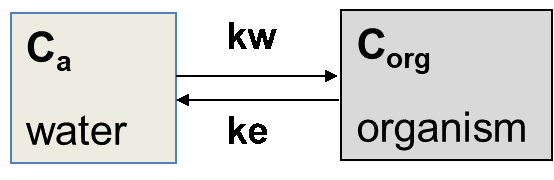

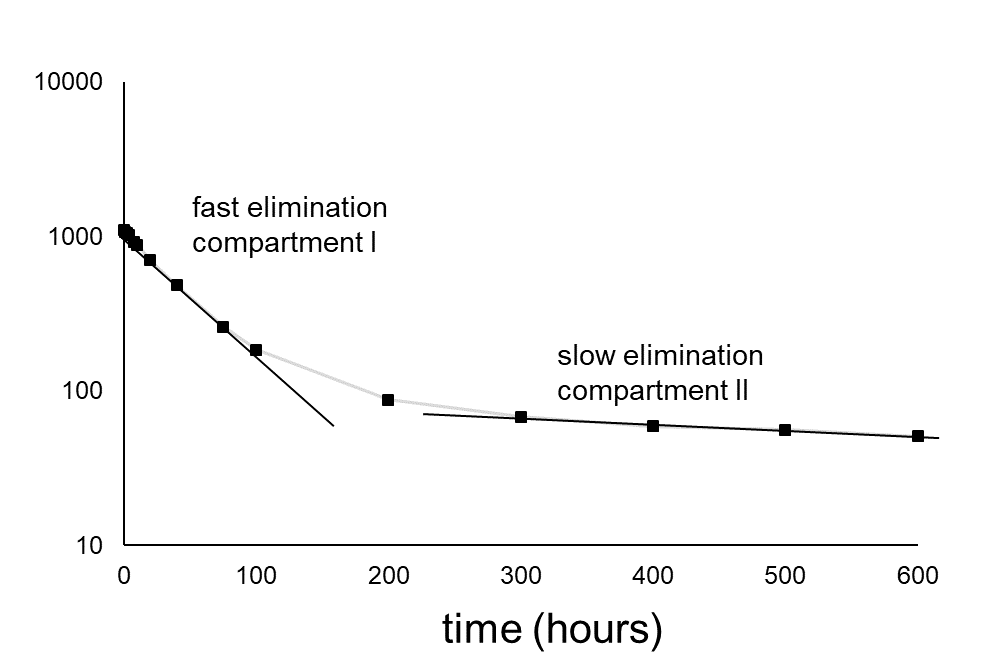
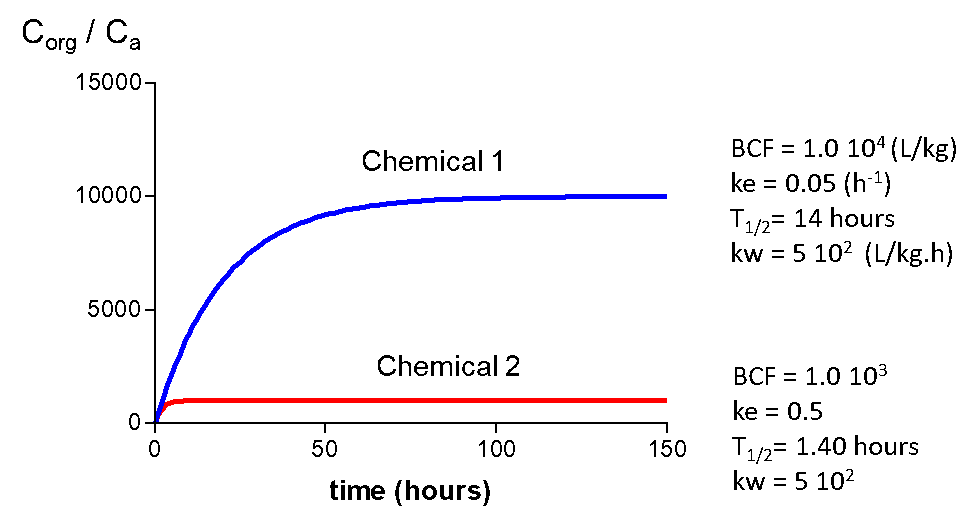
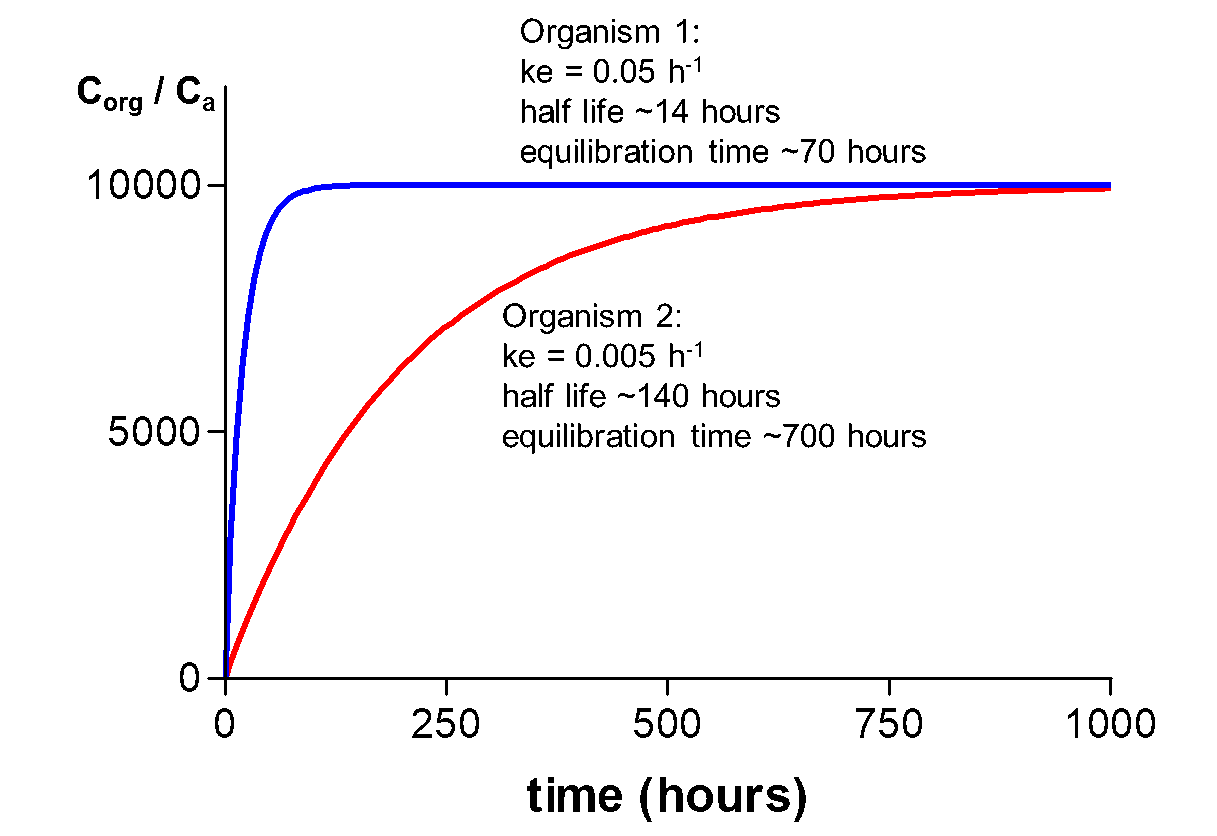
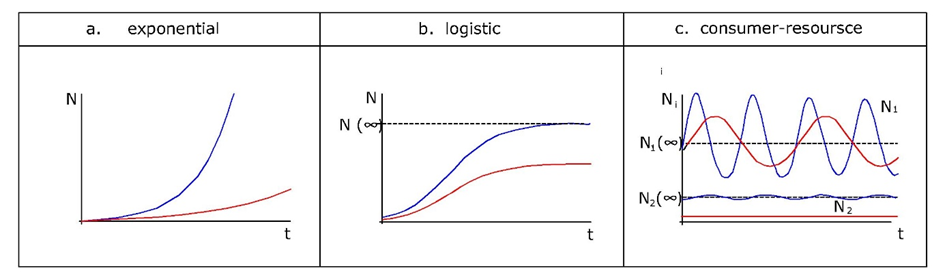
 Corg/Caq becomes constant. This is the steady state situation. The constant Corg/Caq at steady state is called the bioconcentration factor BCF. Mathematically, the BCF can also be calculated from
Corg/Caq becomes constant. This is the steady state situation. The constant Corg/Caq at steady state is called the bioconcentration factor BCF. Mathematically, the BCF can also be calculated from  kw/ke. This follows directly from equation 4: after long exposure time (t),
kw/ke. This follows directly from equation 4: after long exposure time (t), 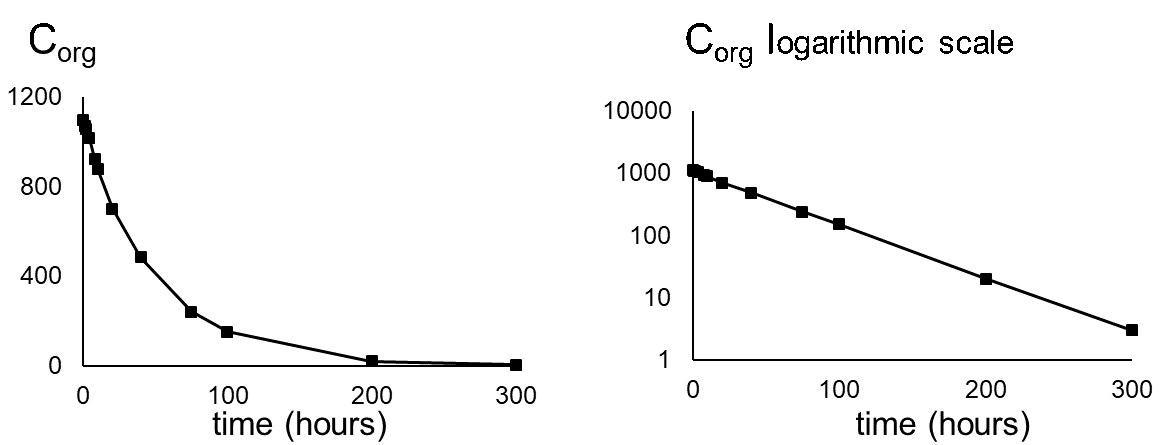
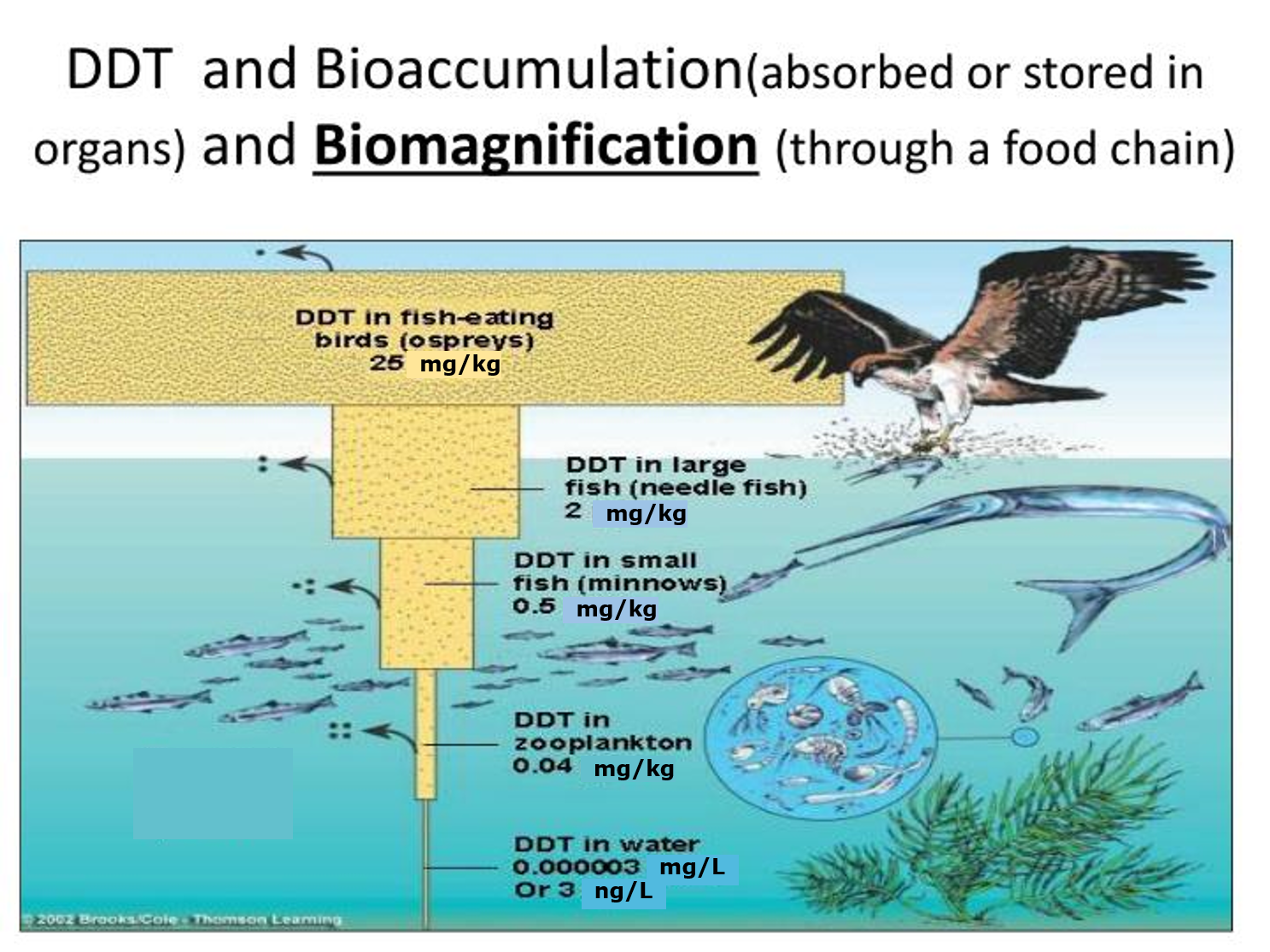
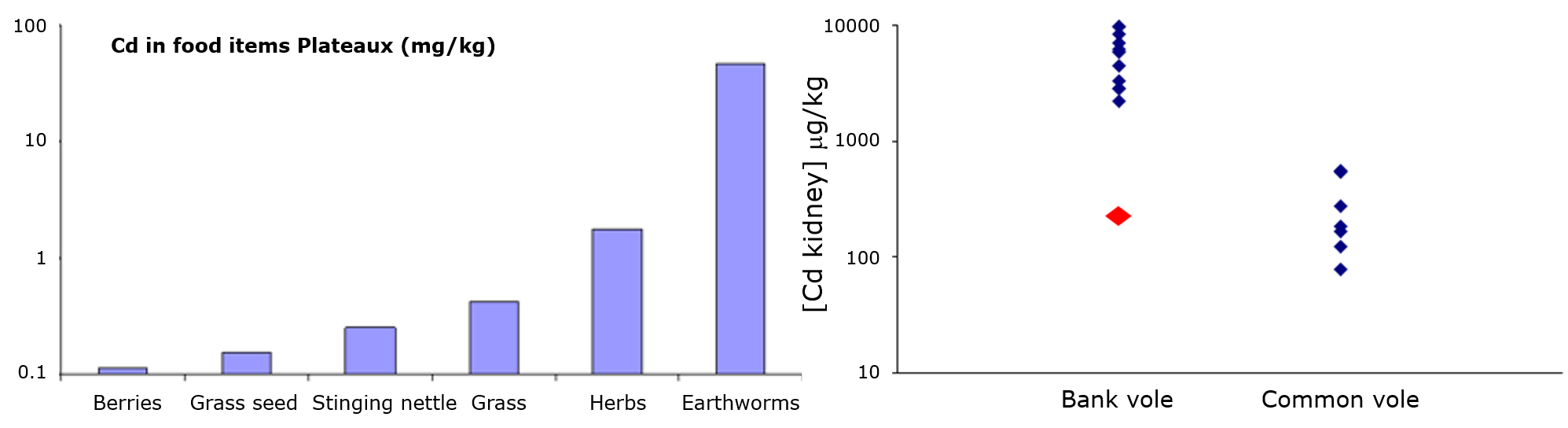
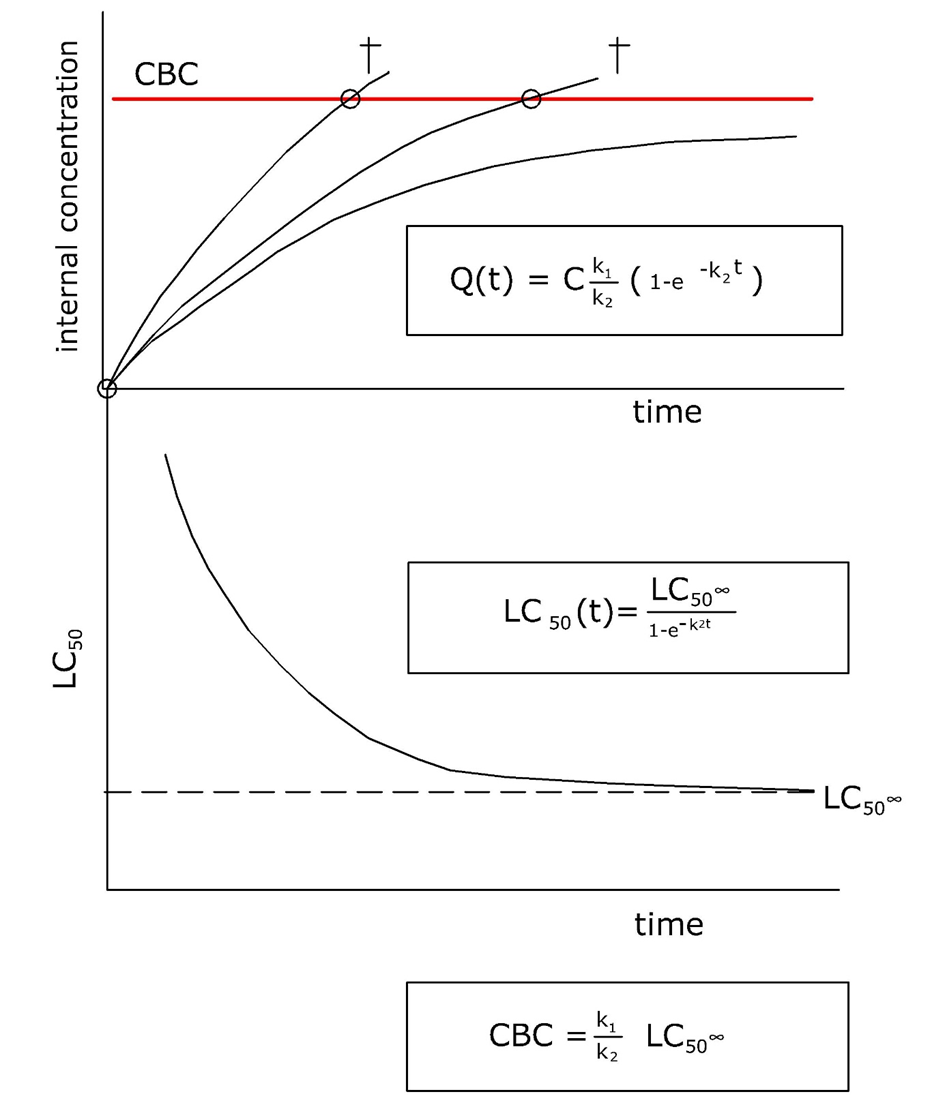

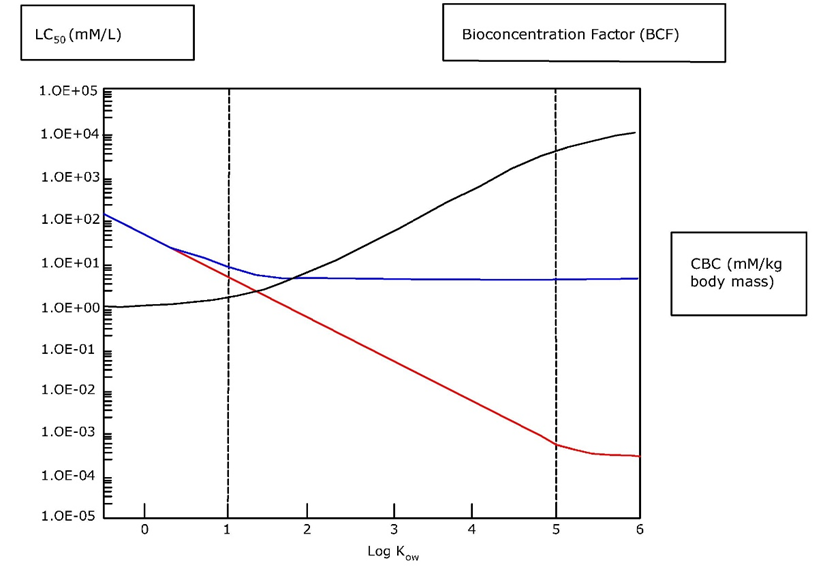

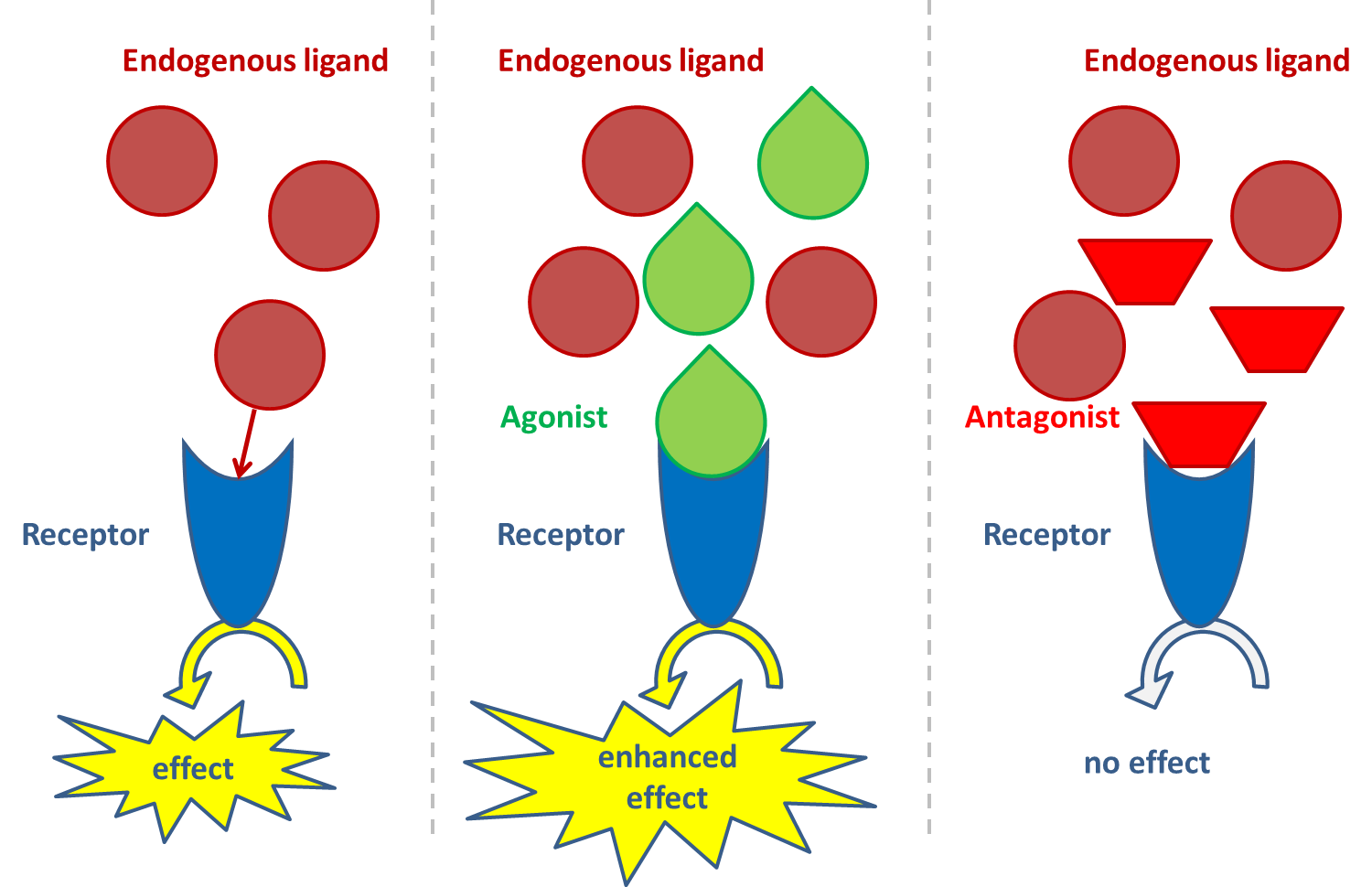
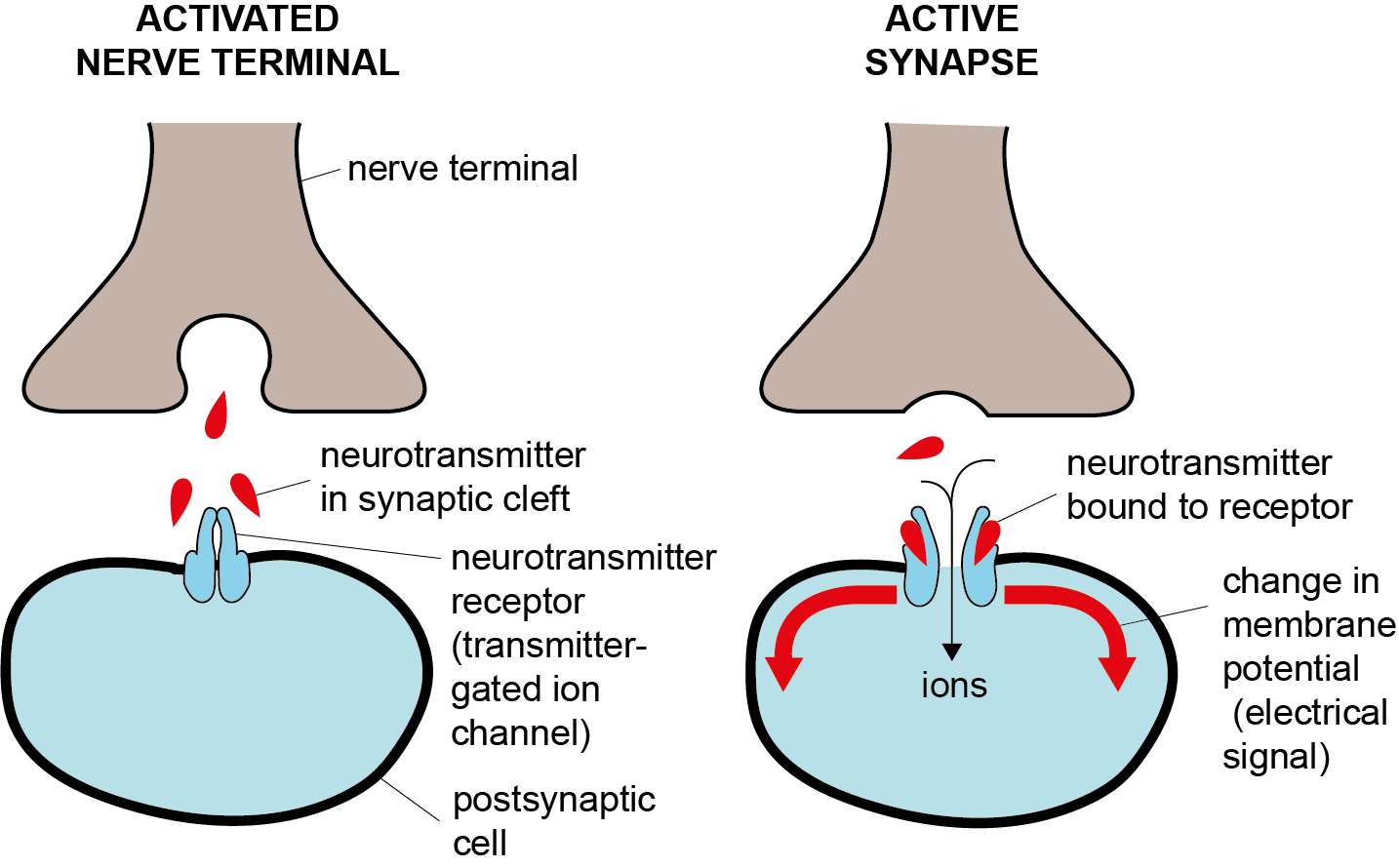
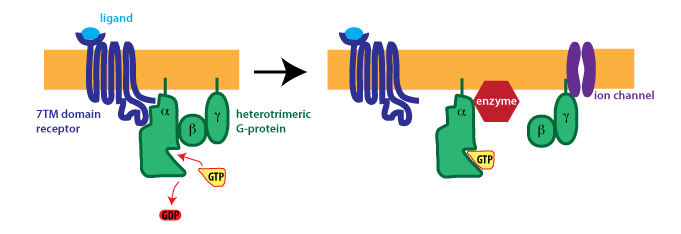
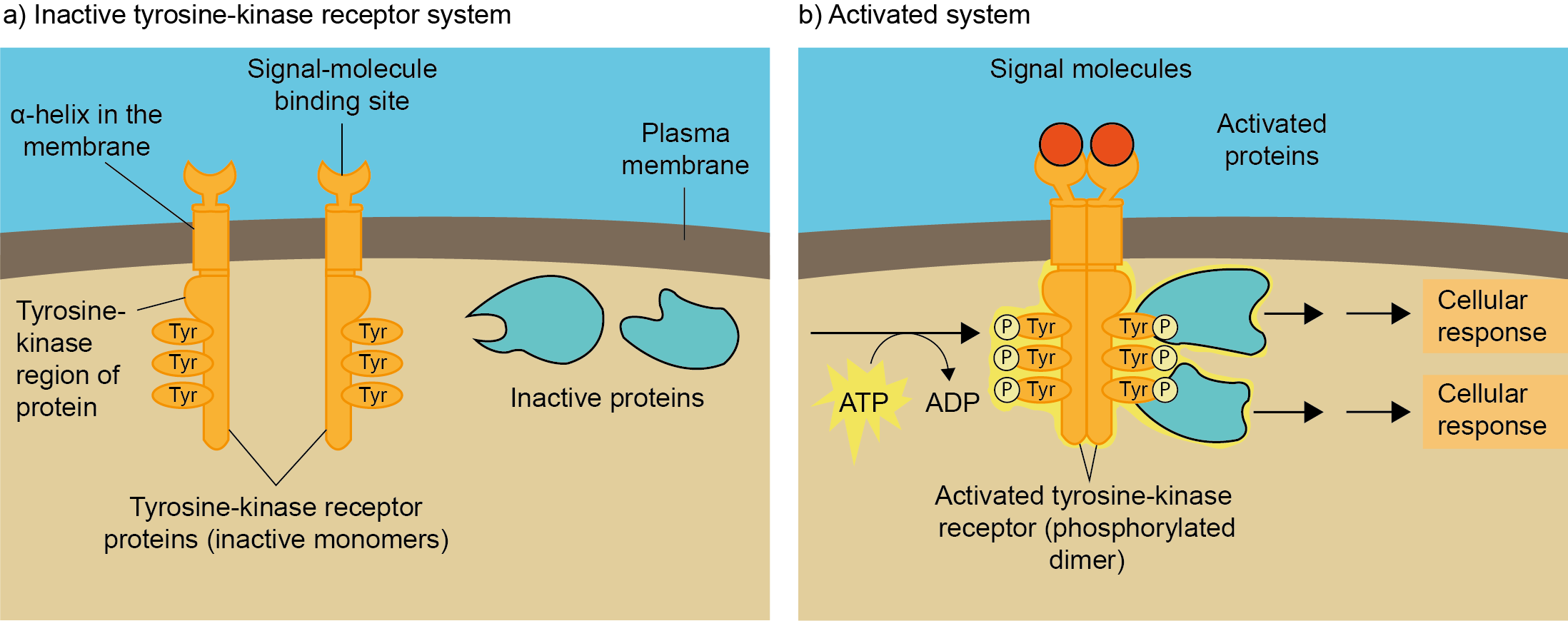
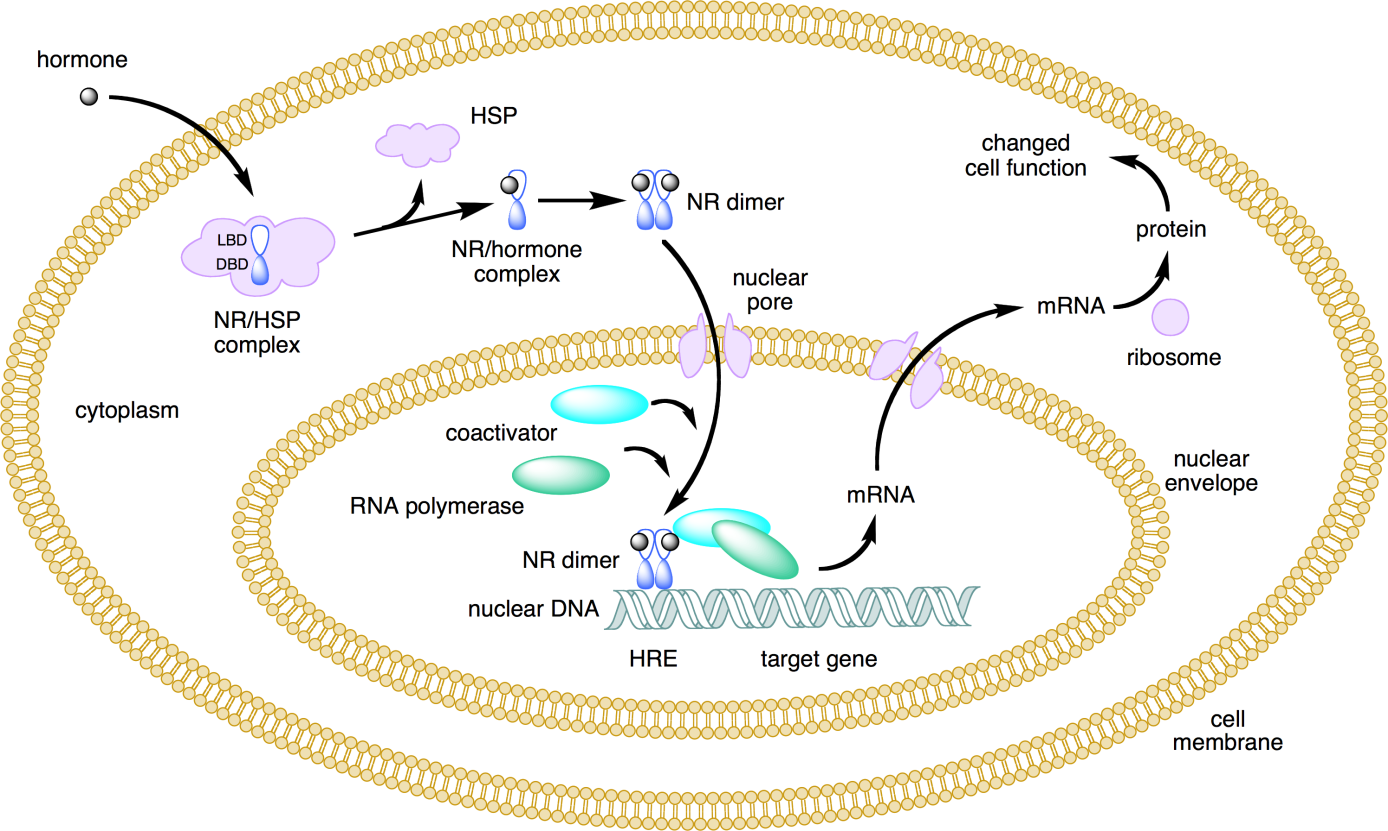
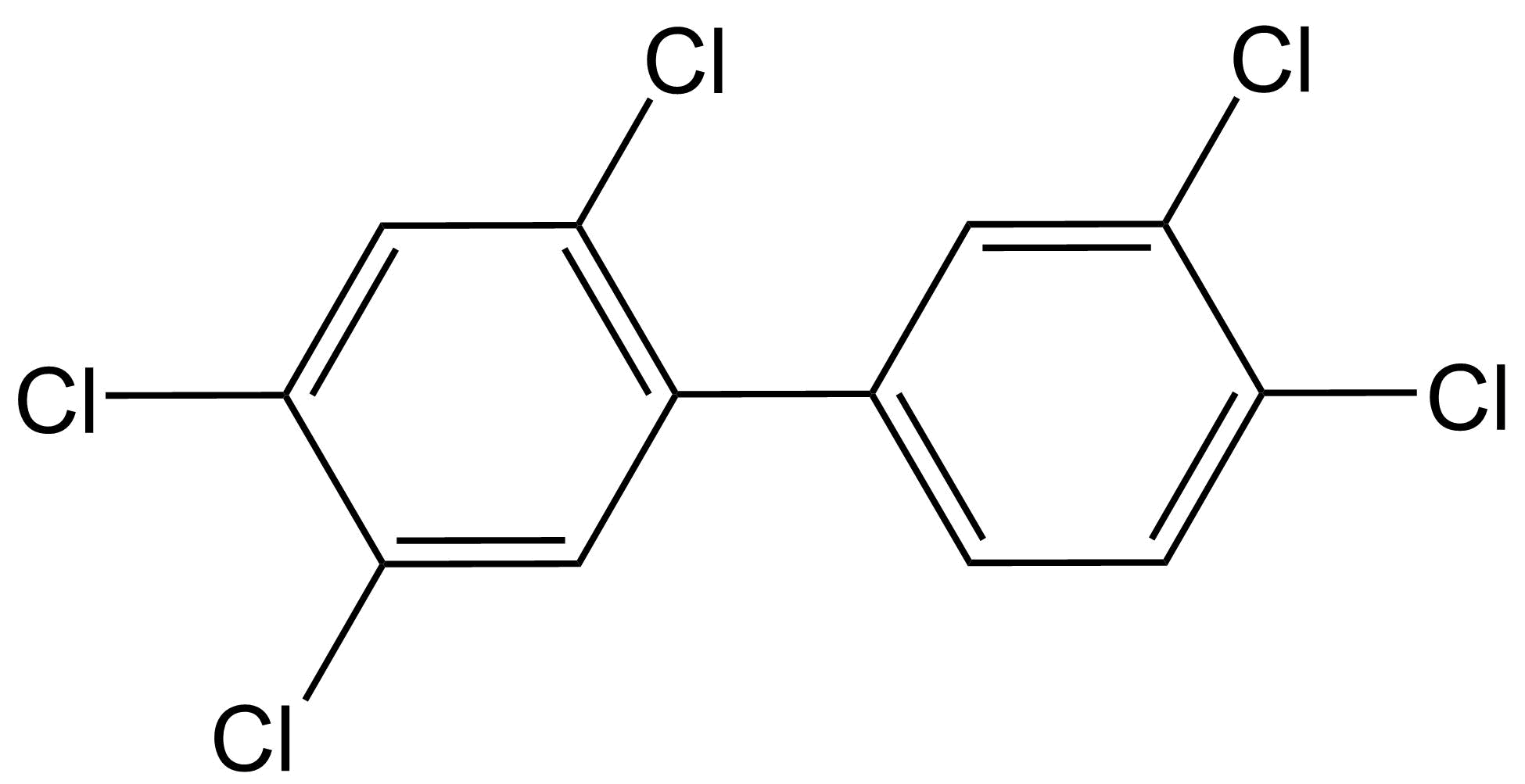
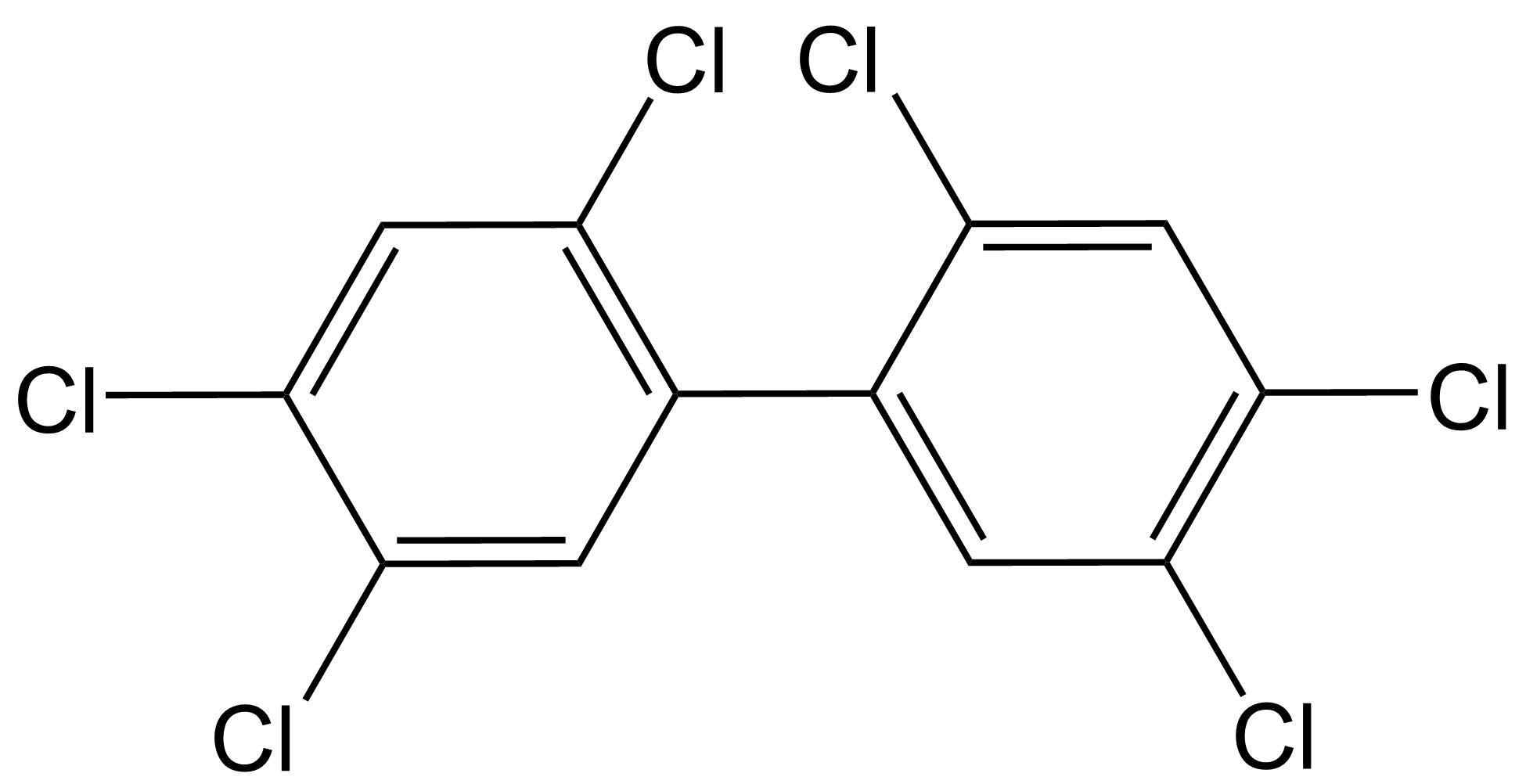
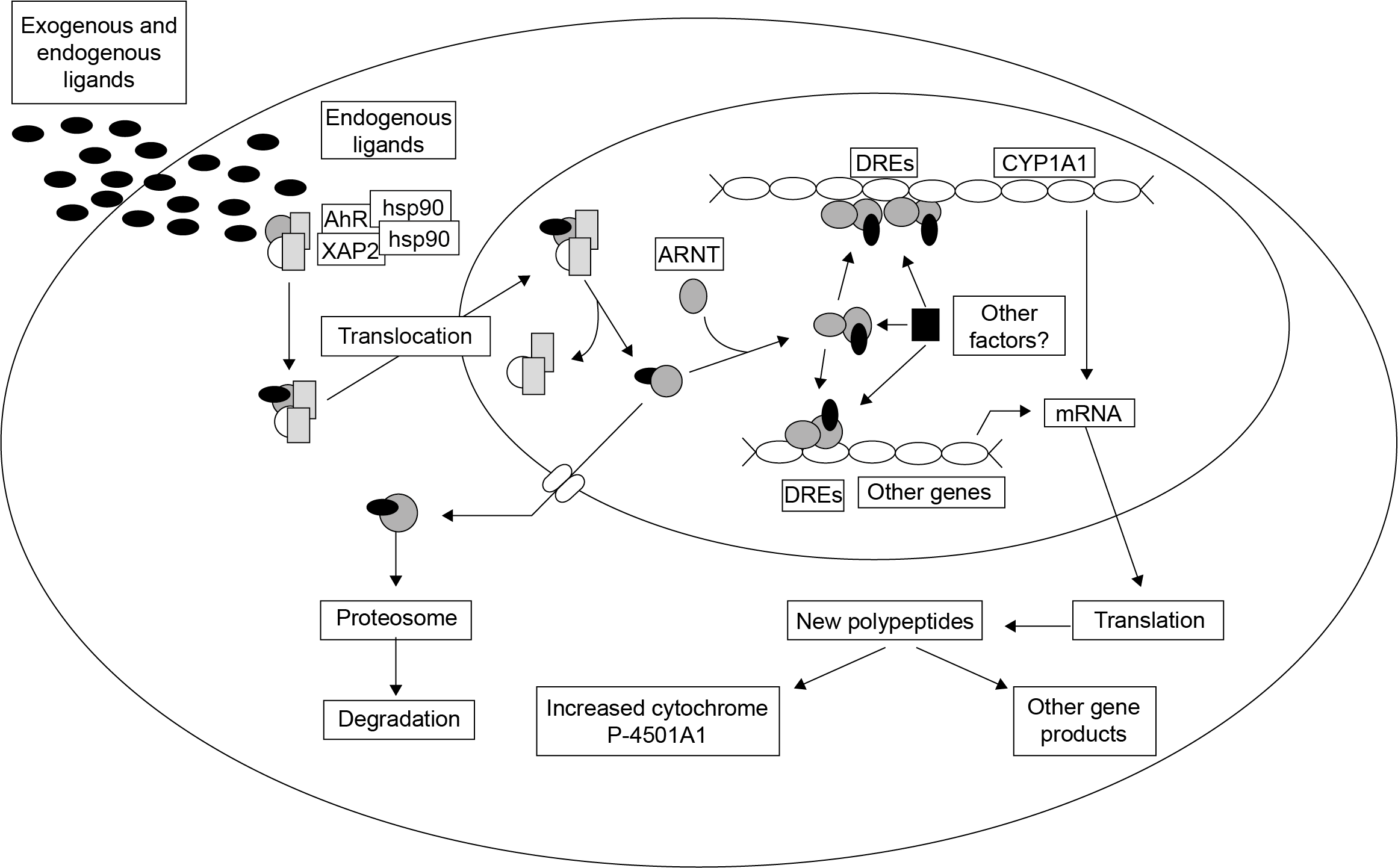
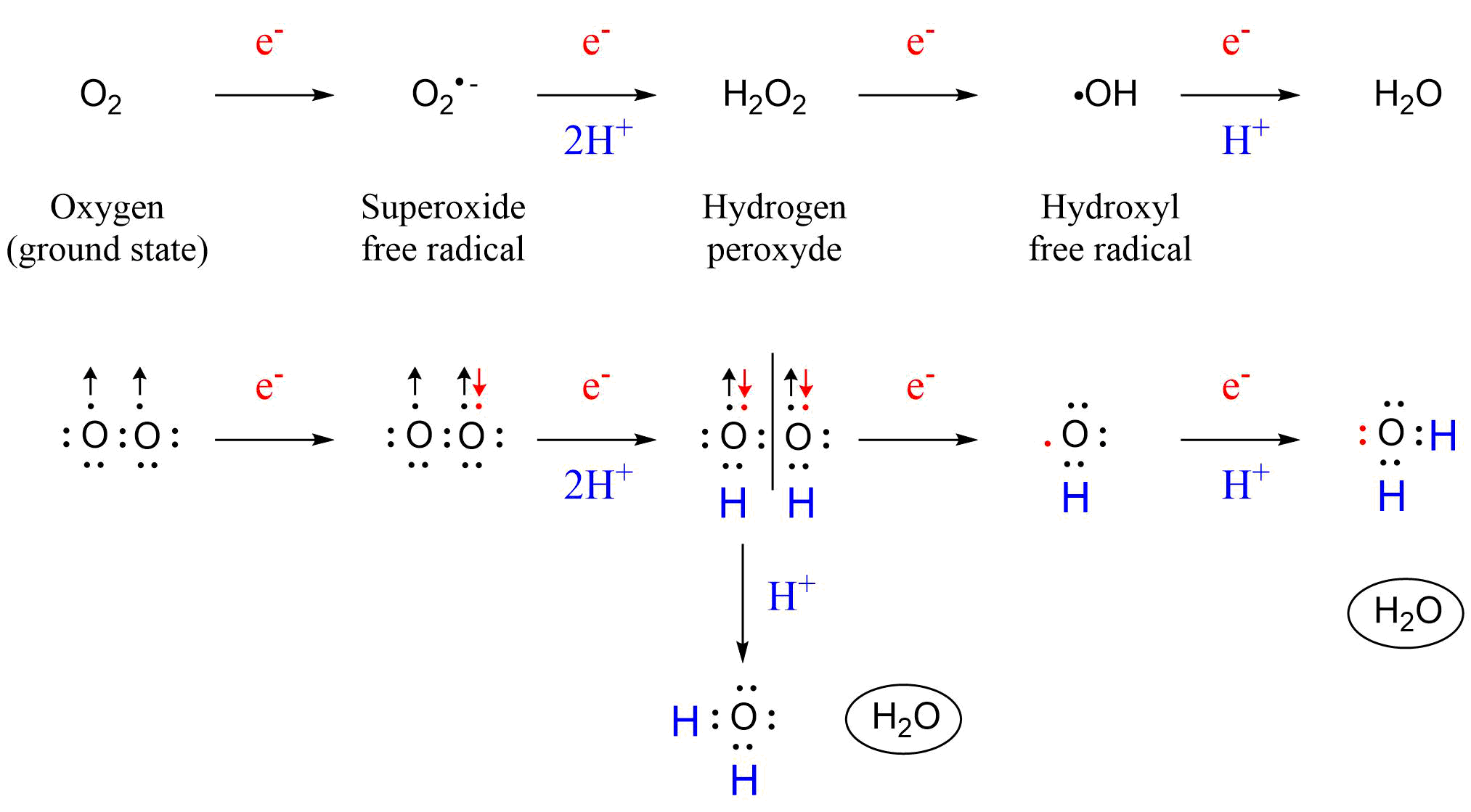
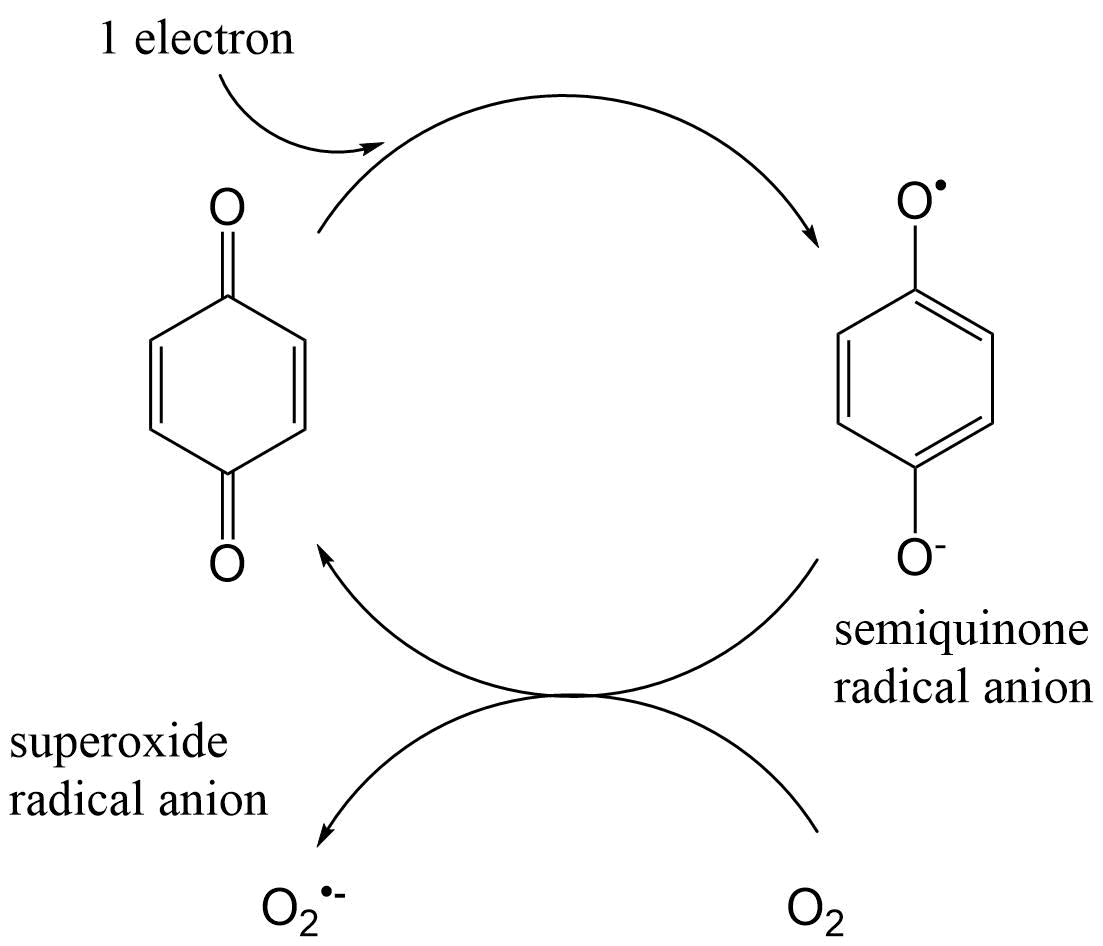
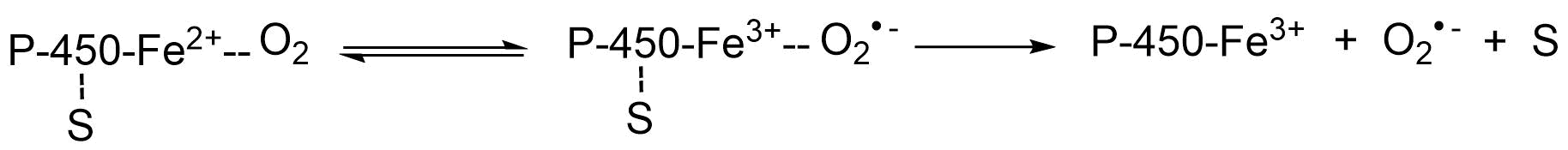
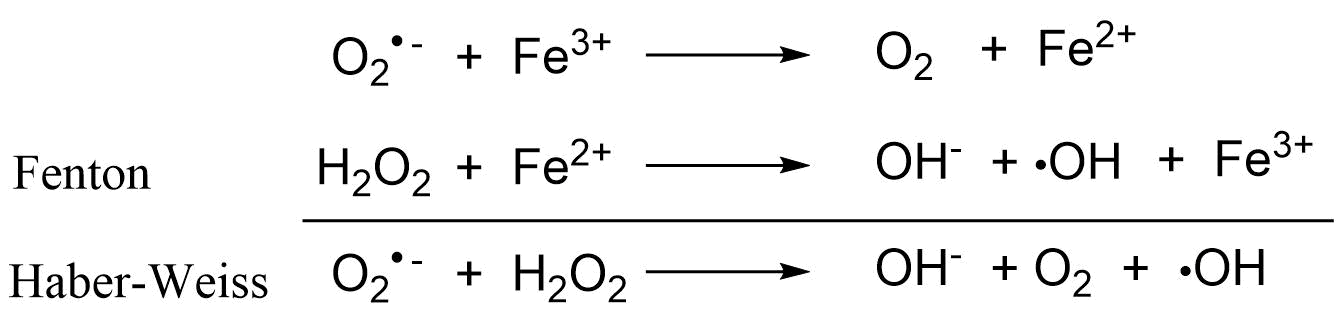
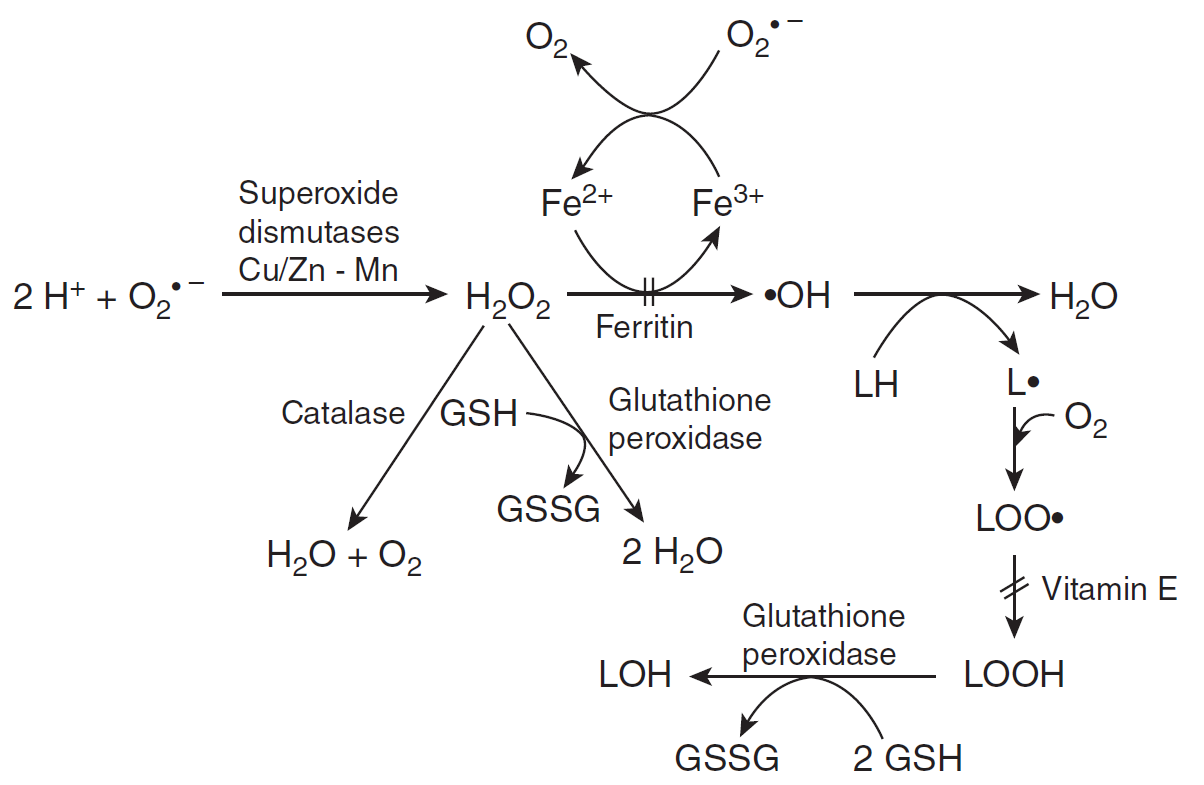
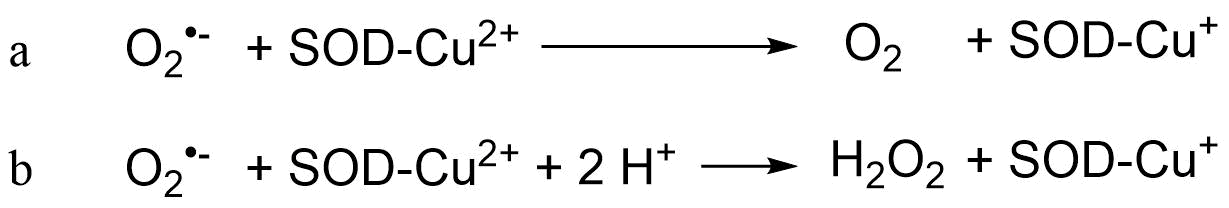
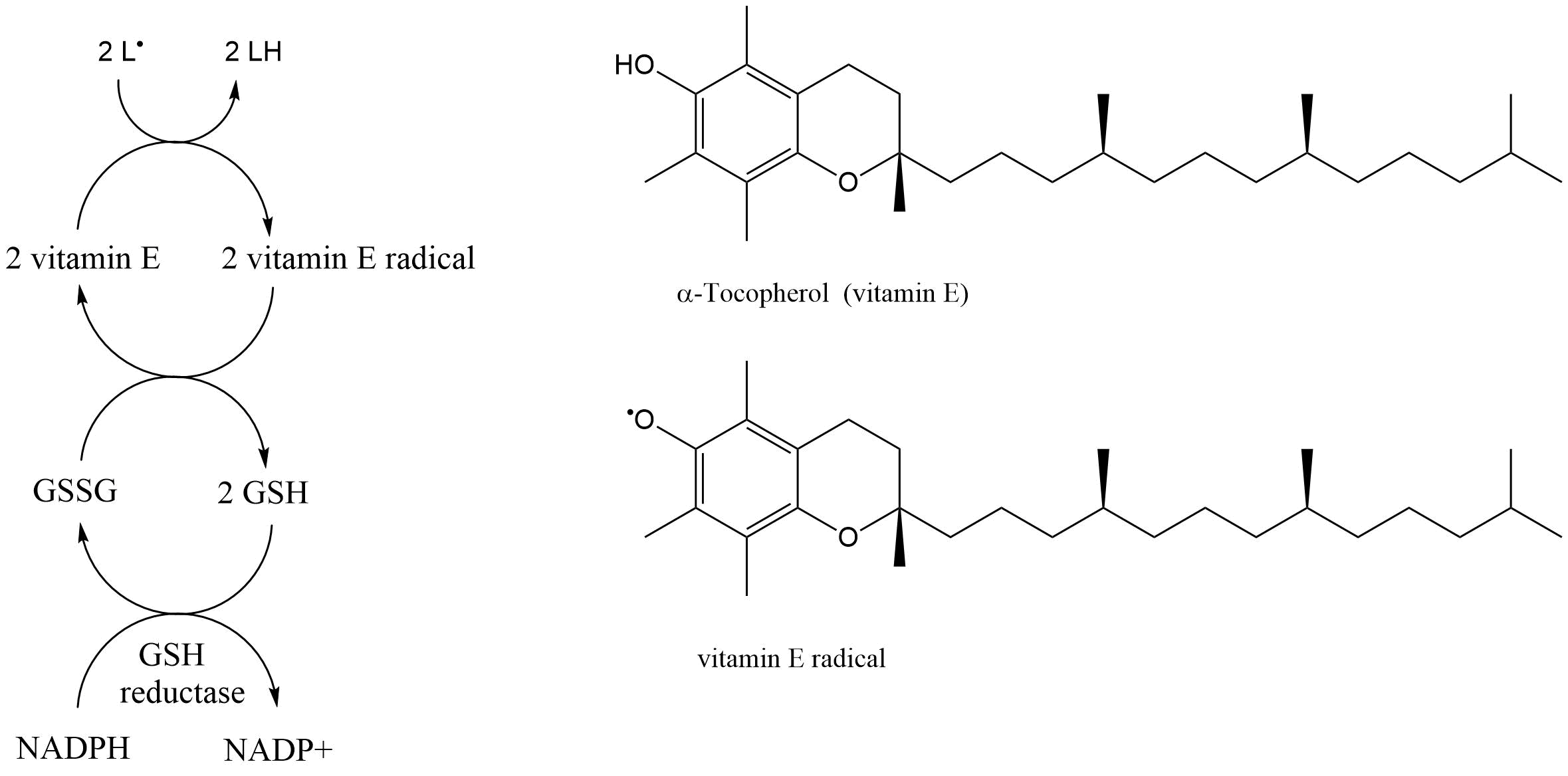
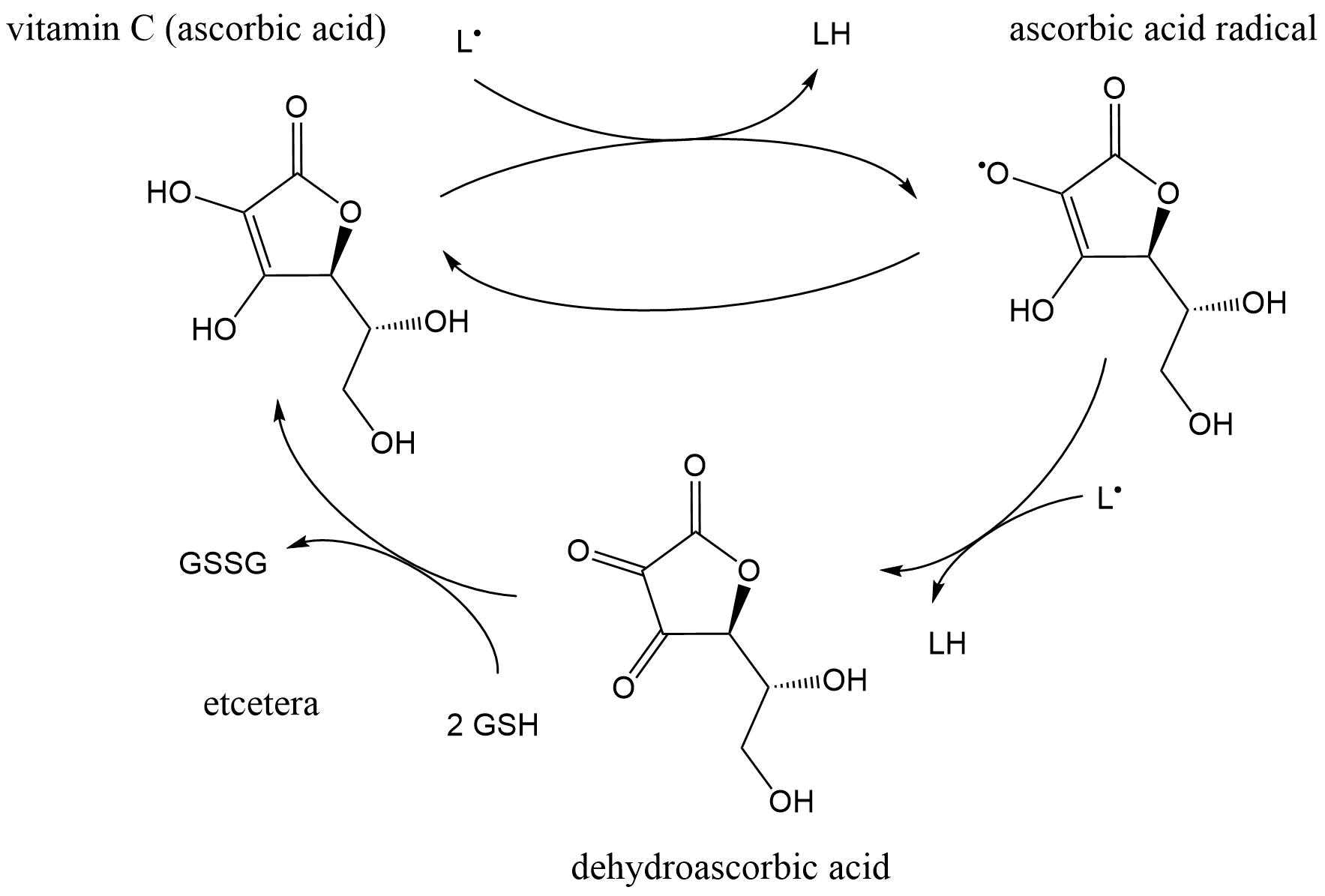
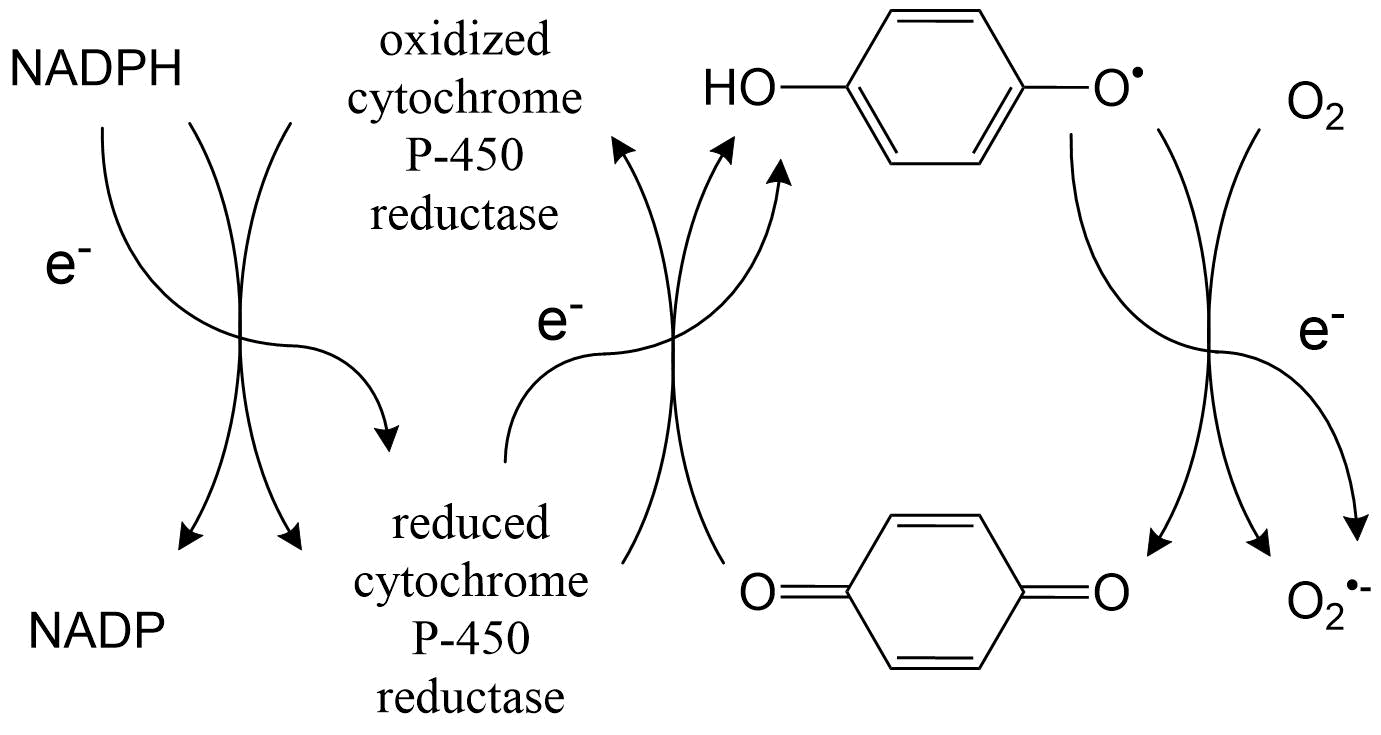
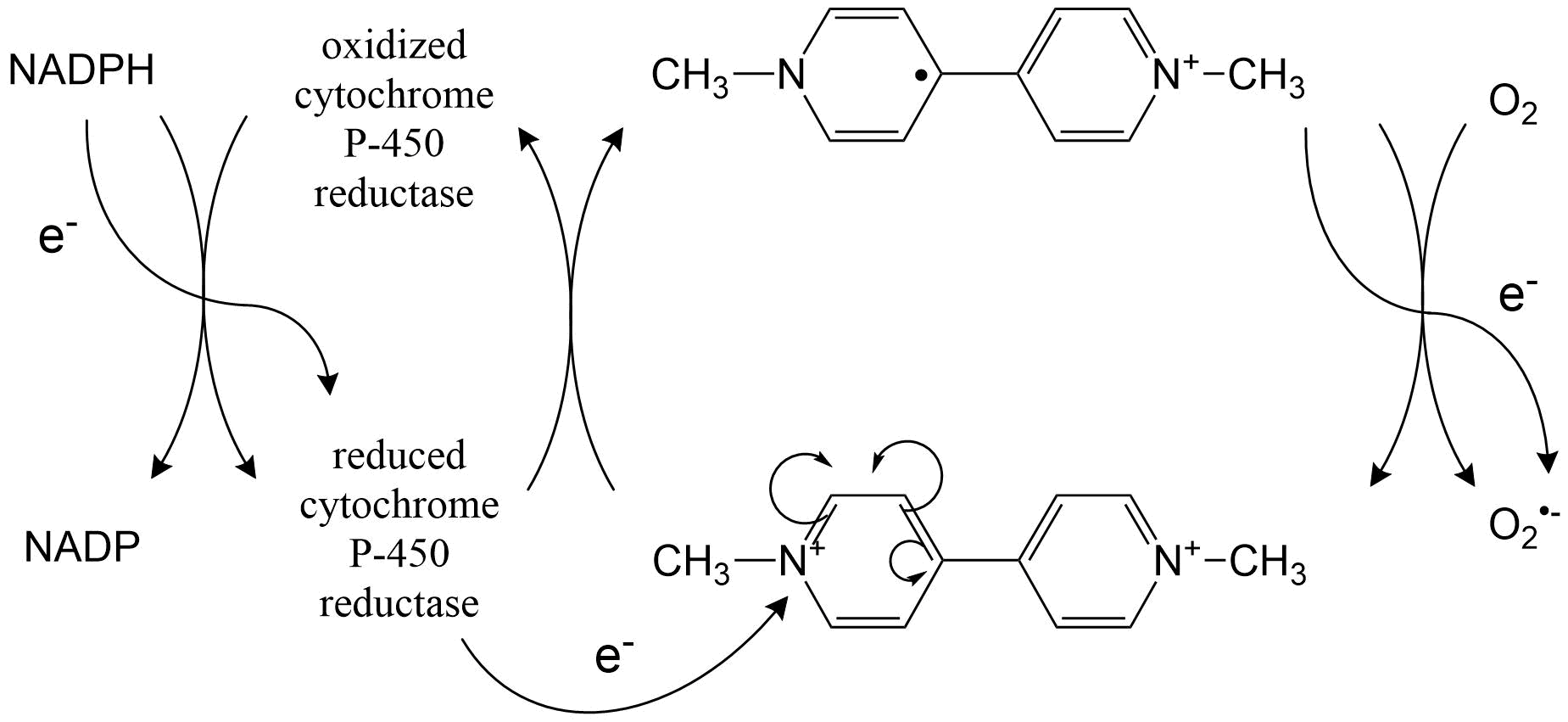
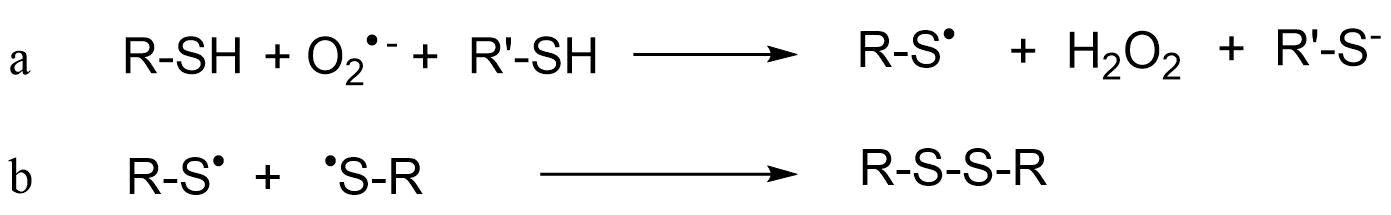
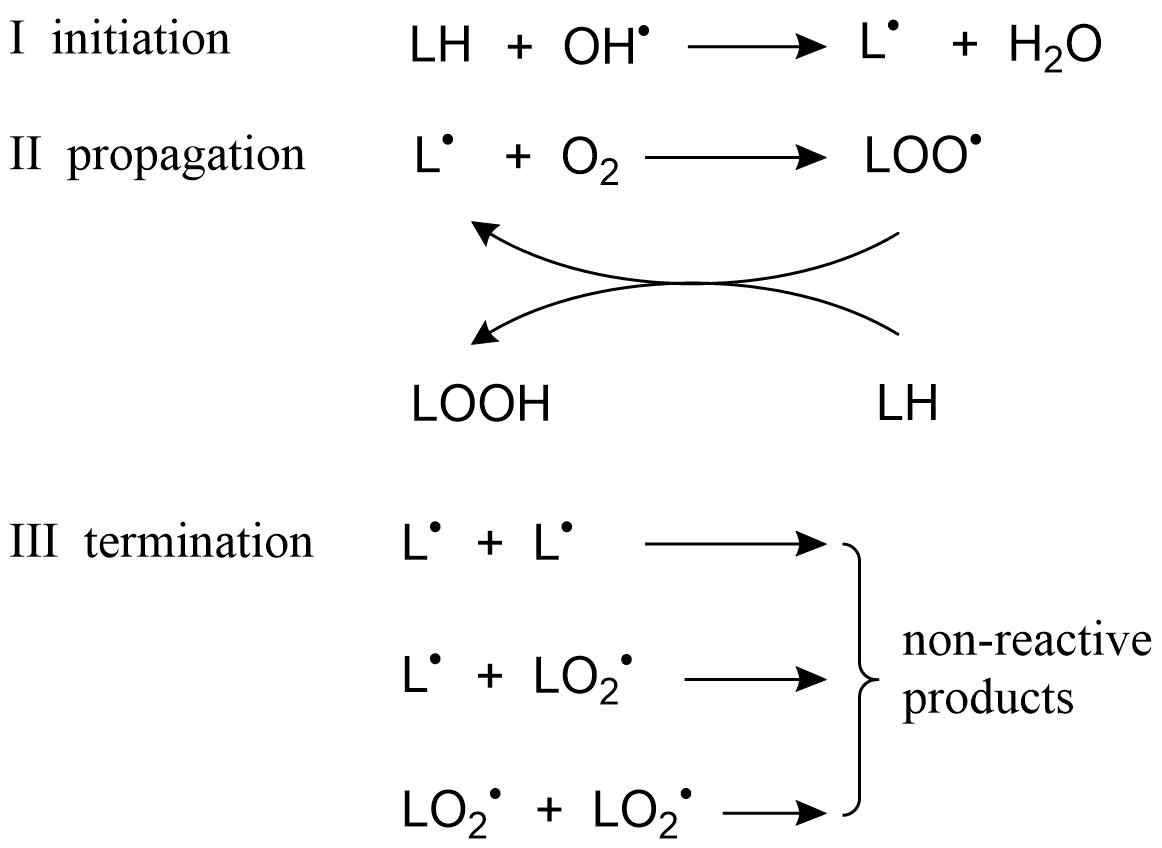
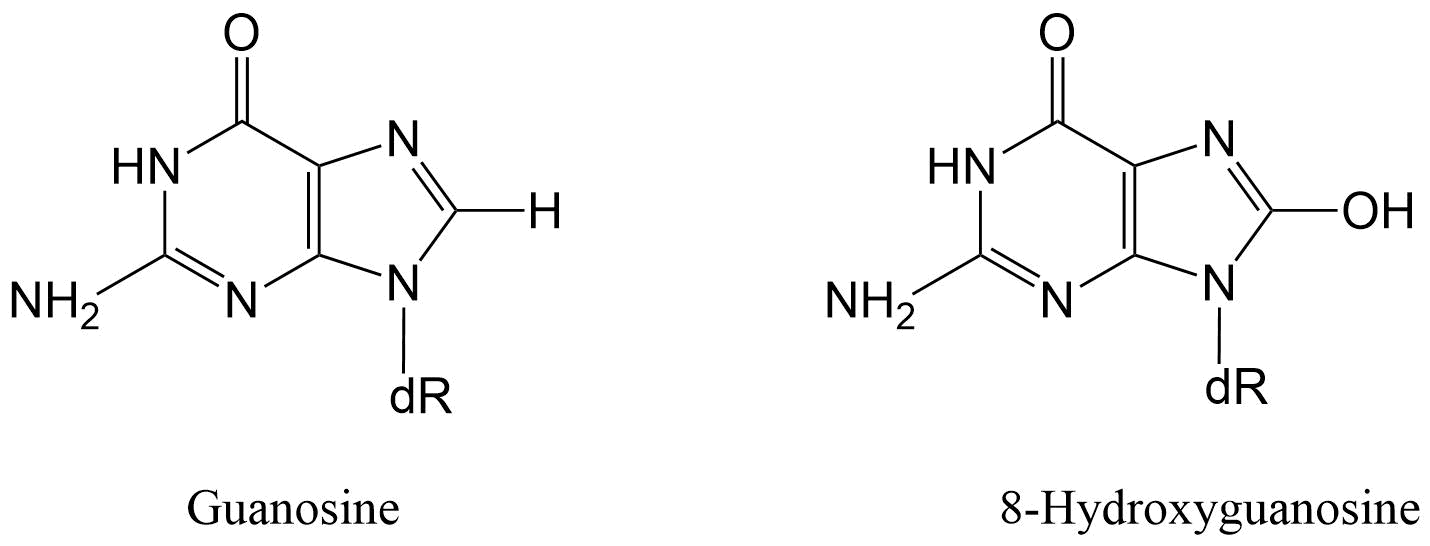







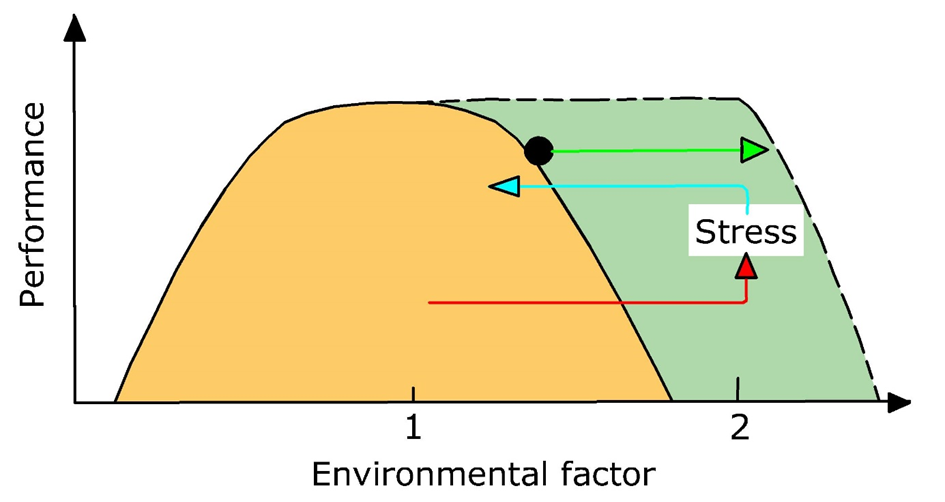

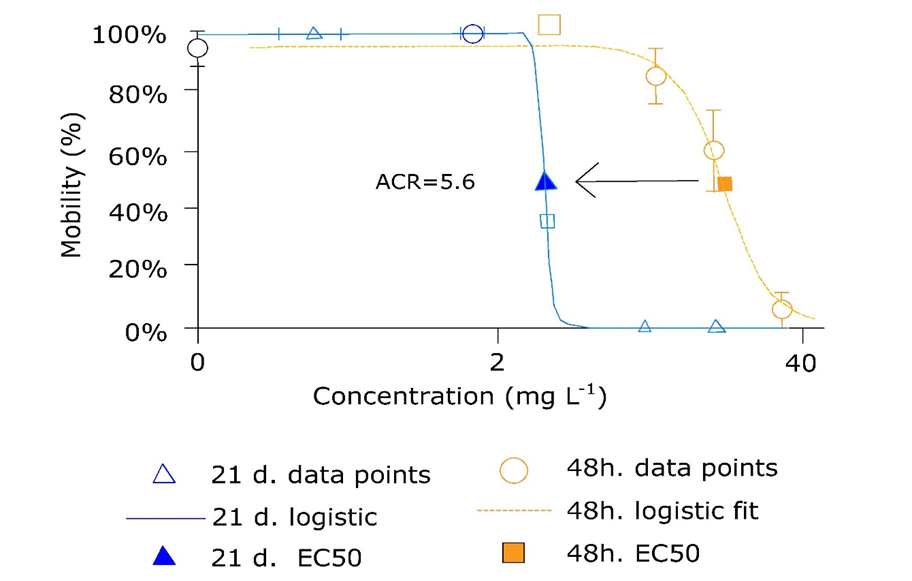
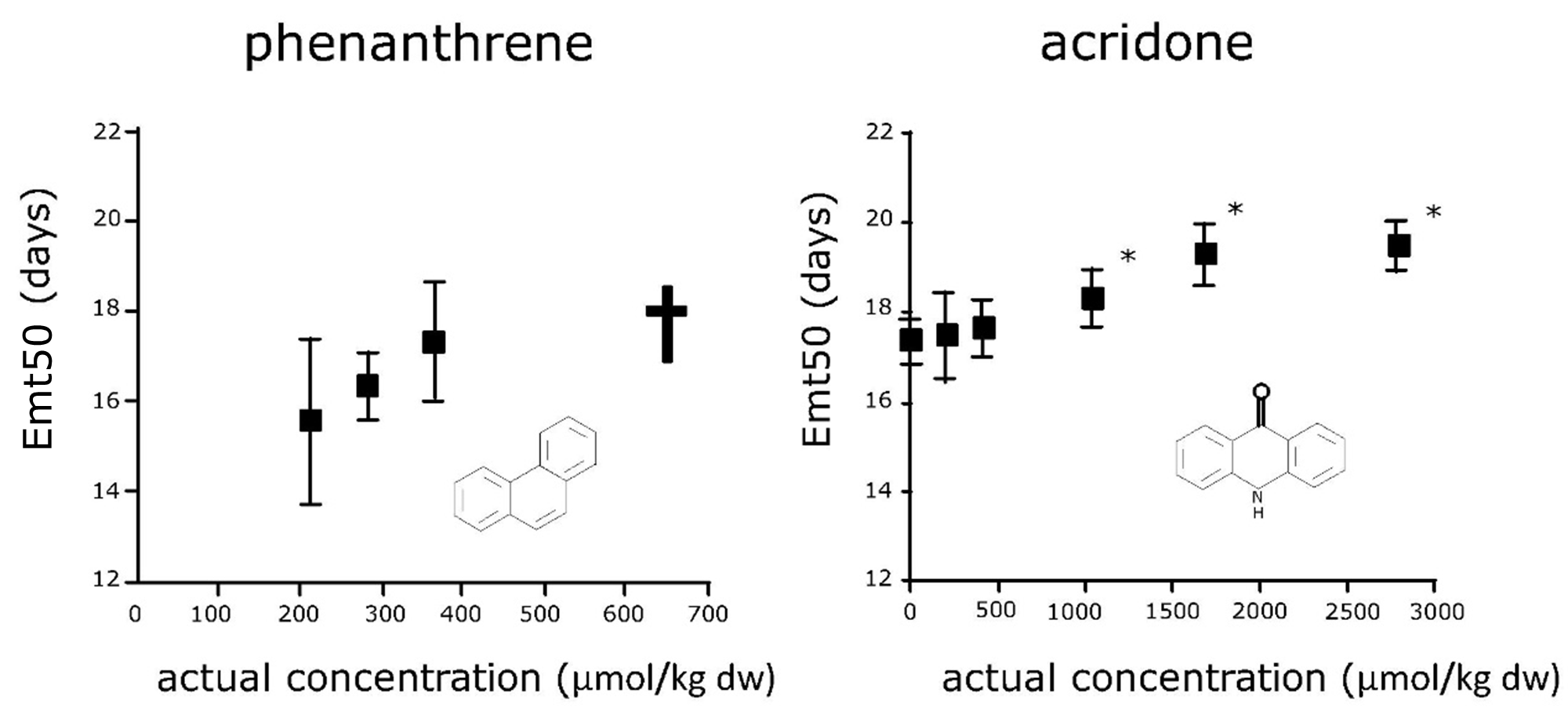
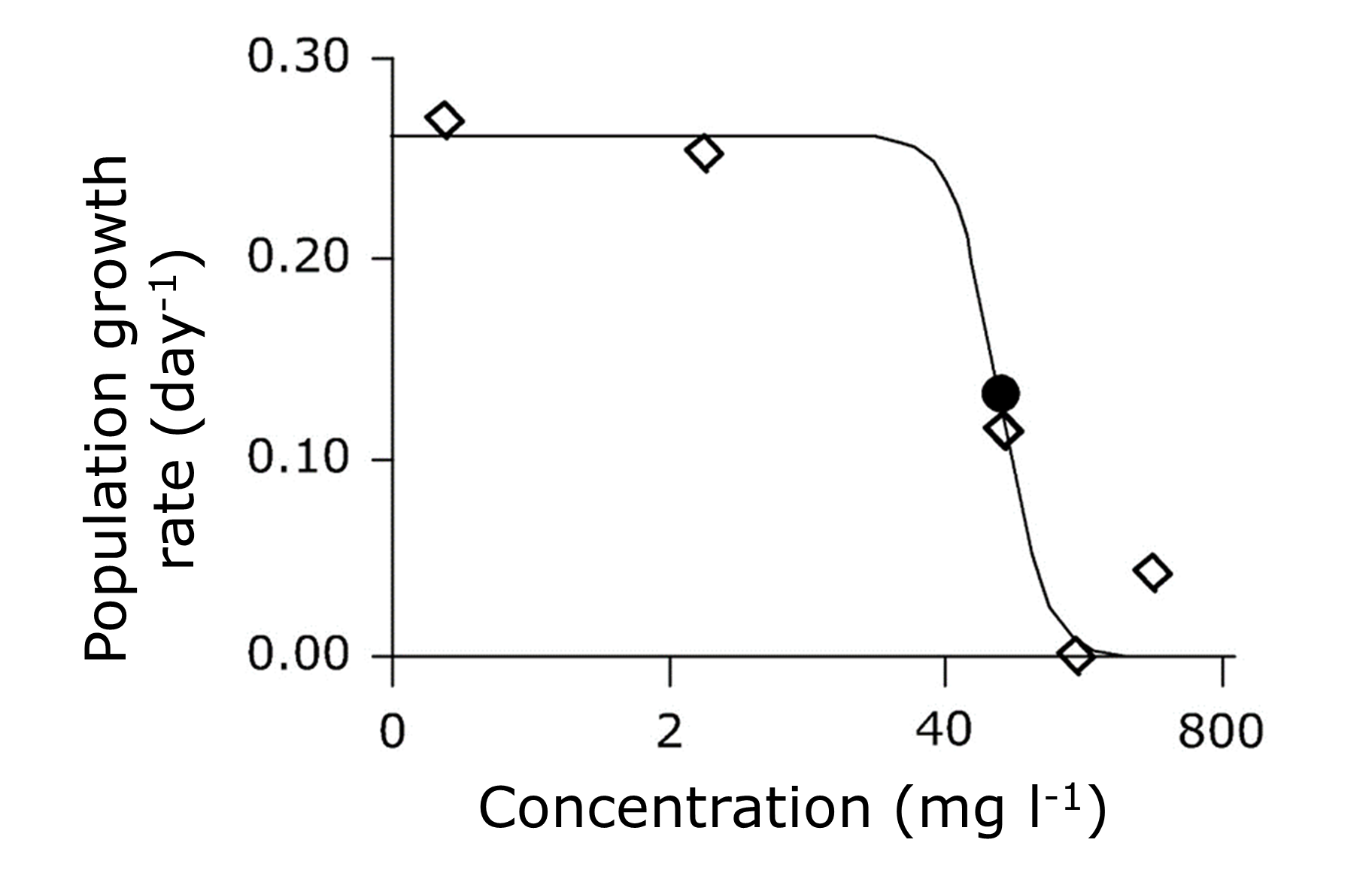

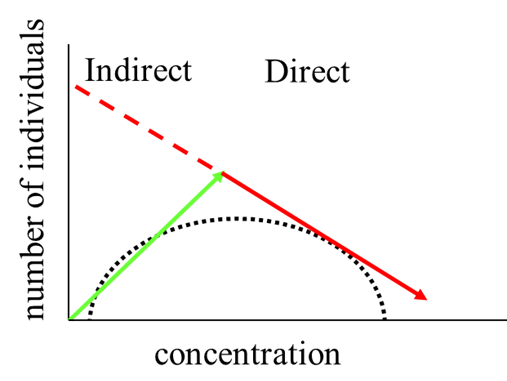

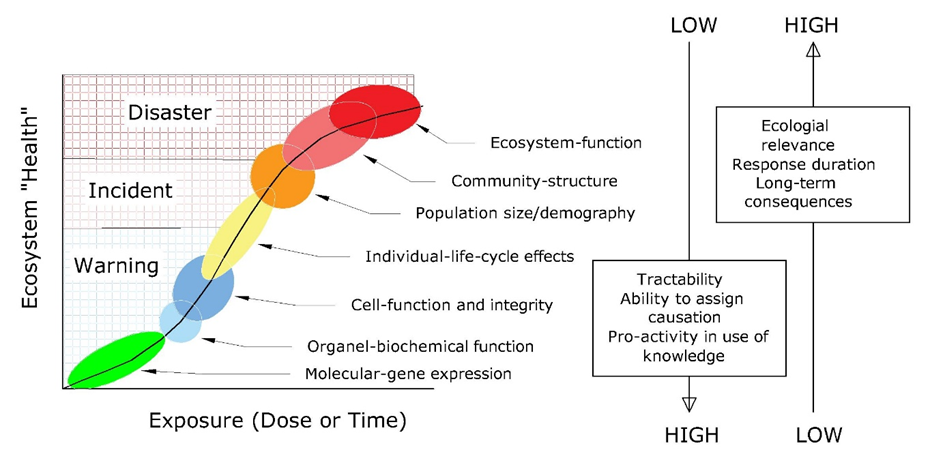

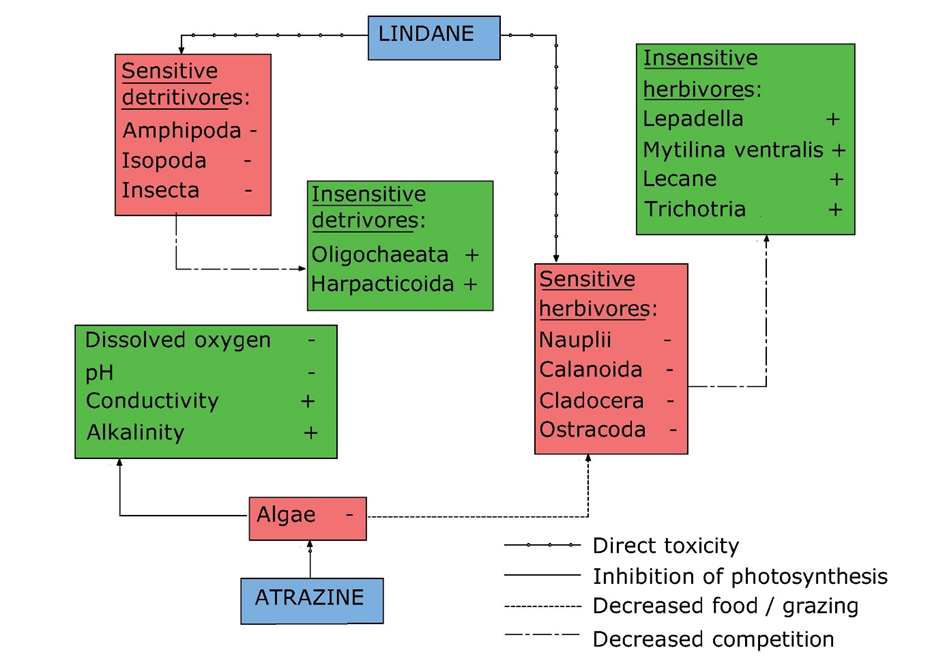
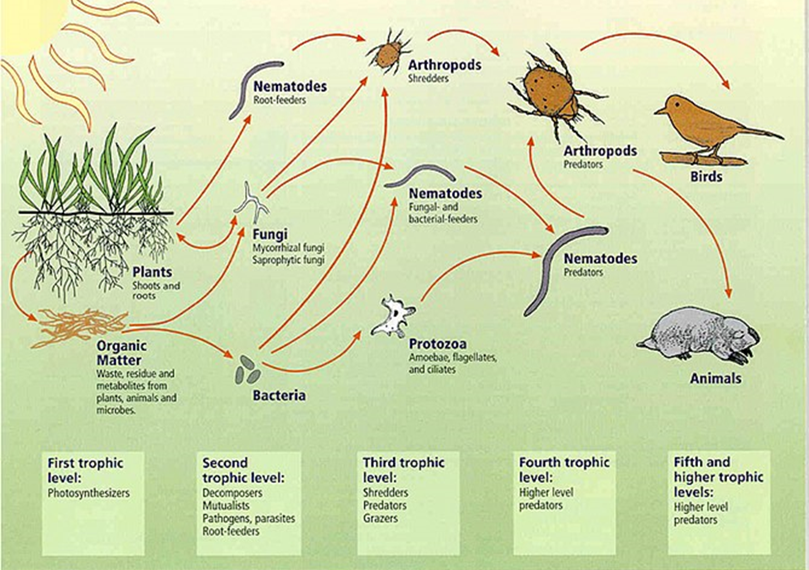
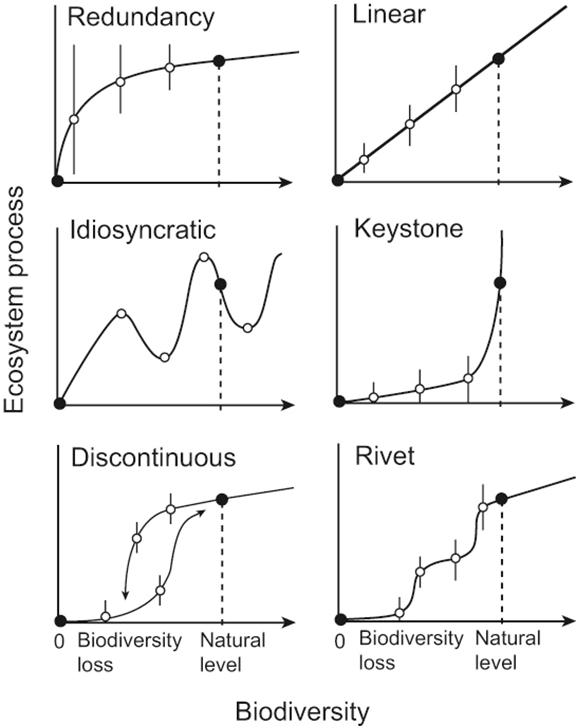
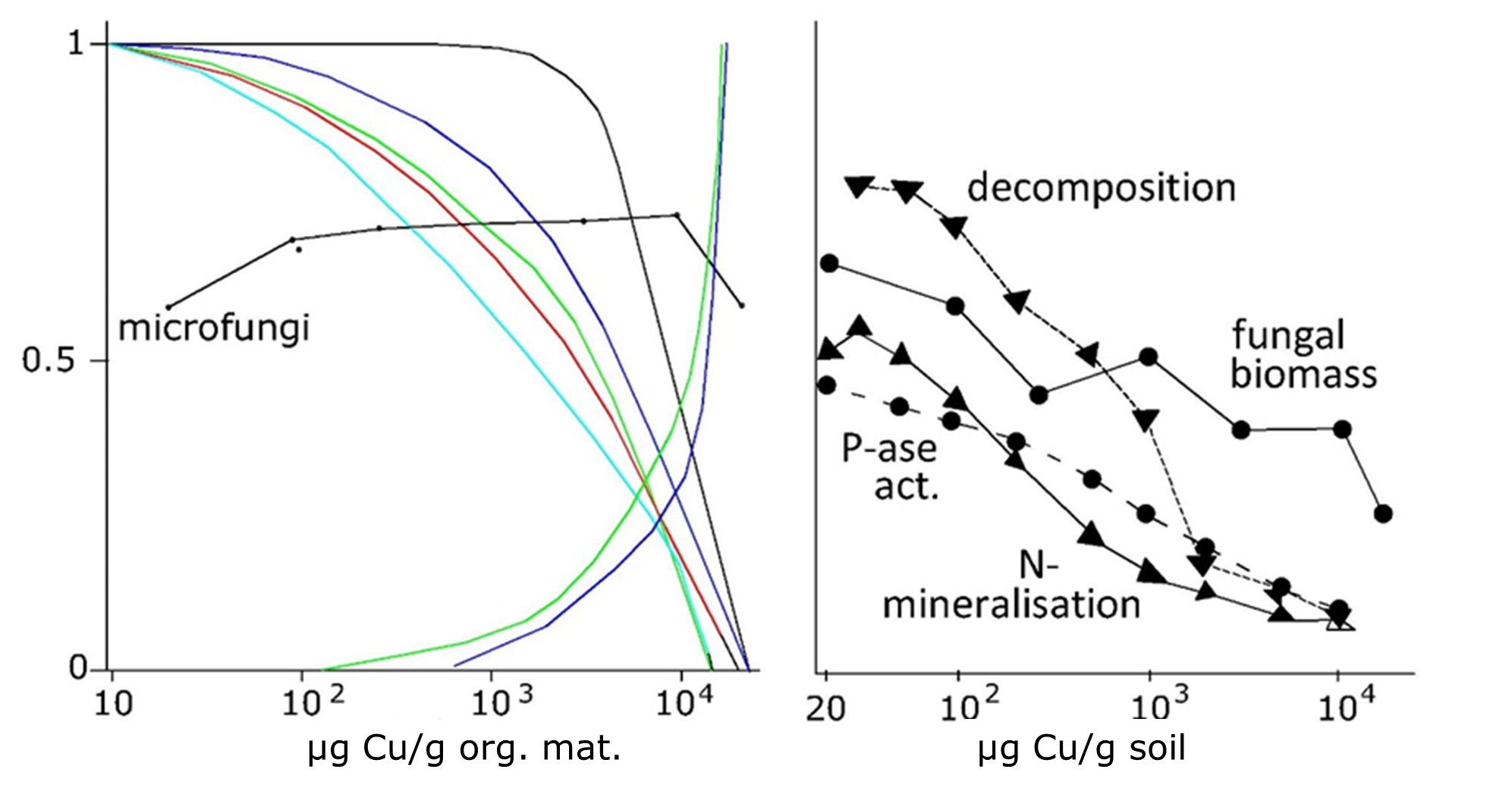
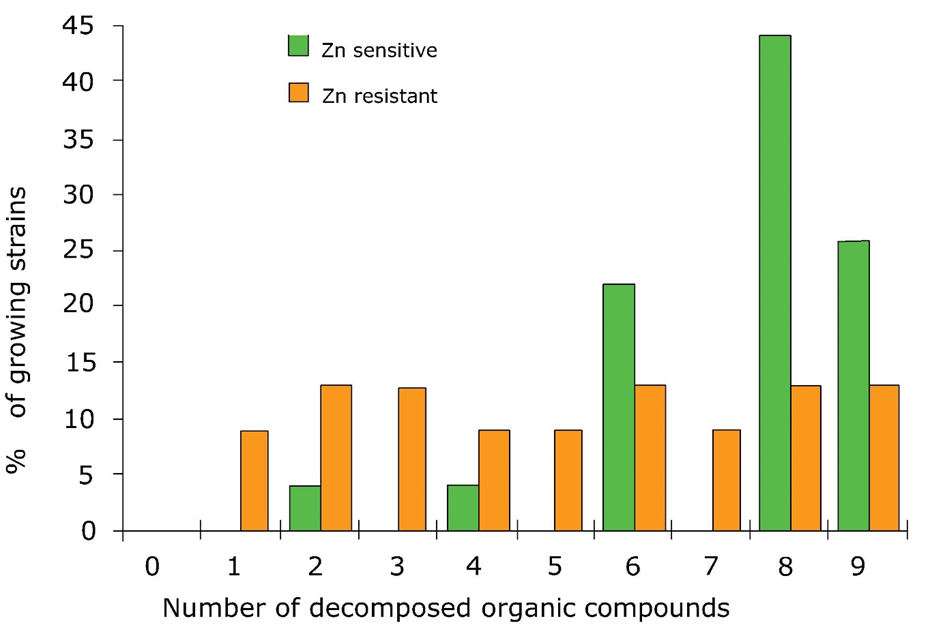
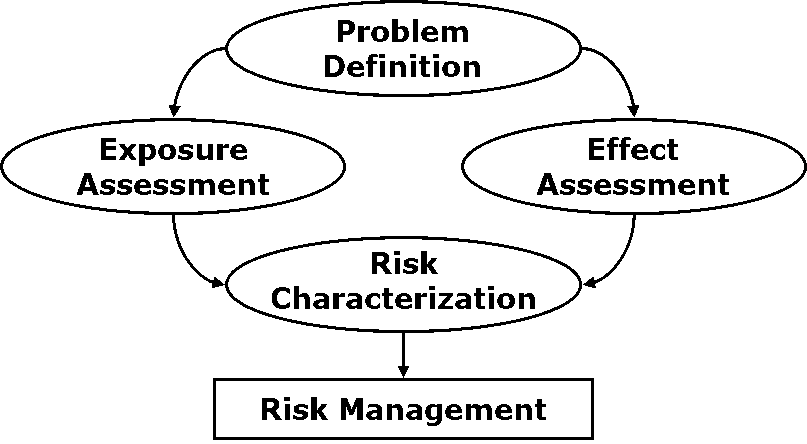


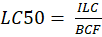 (2)
(2)

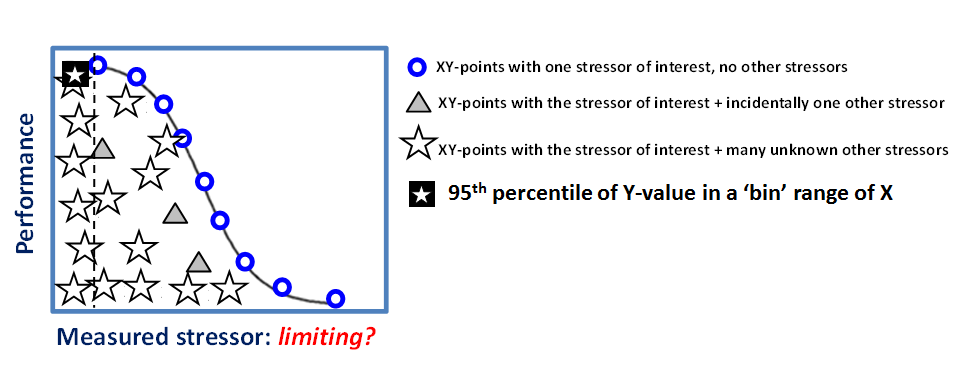

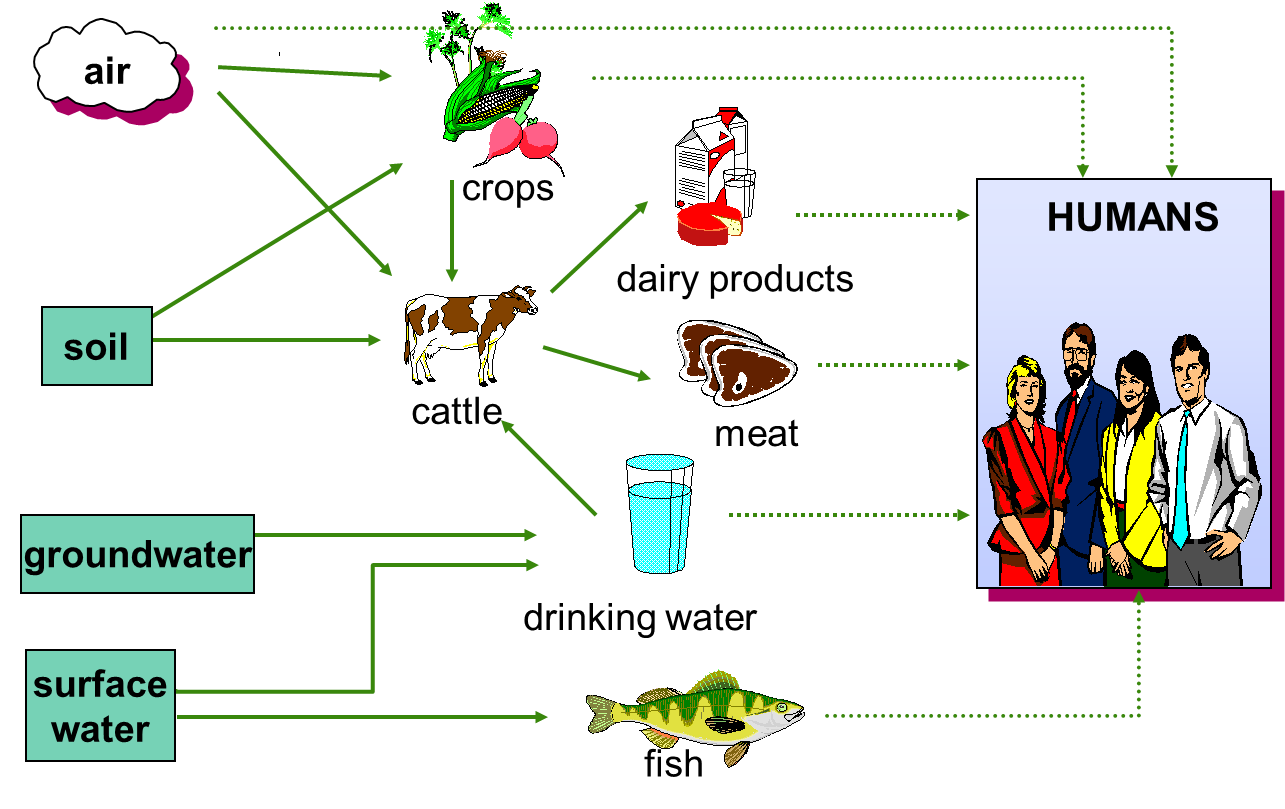
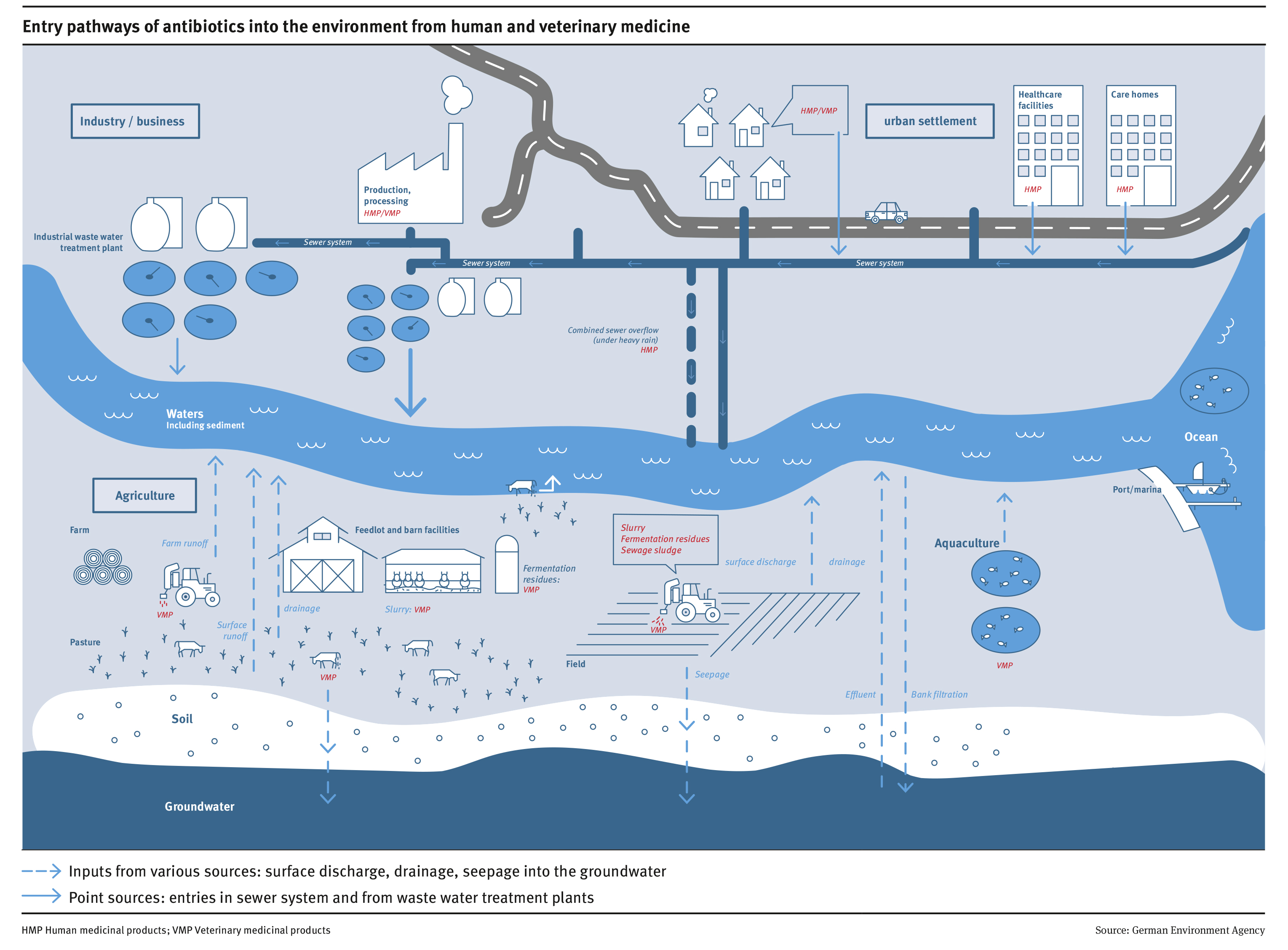
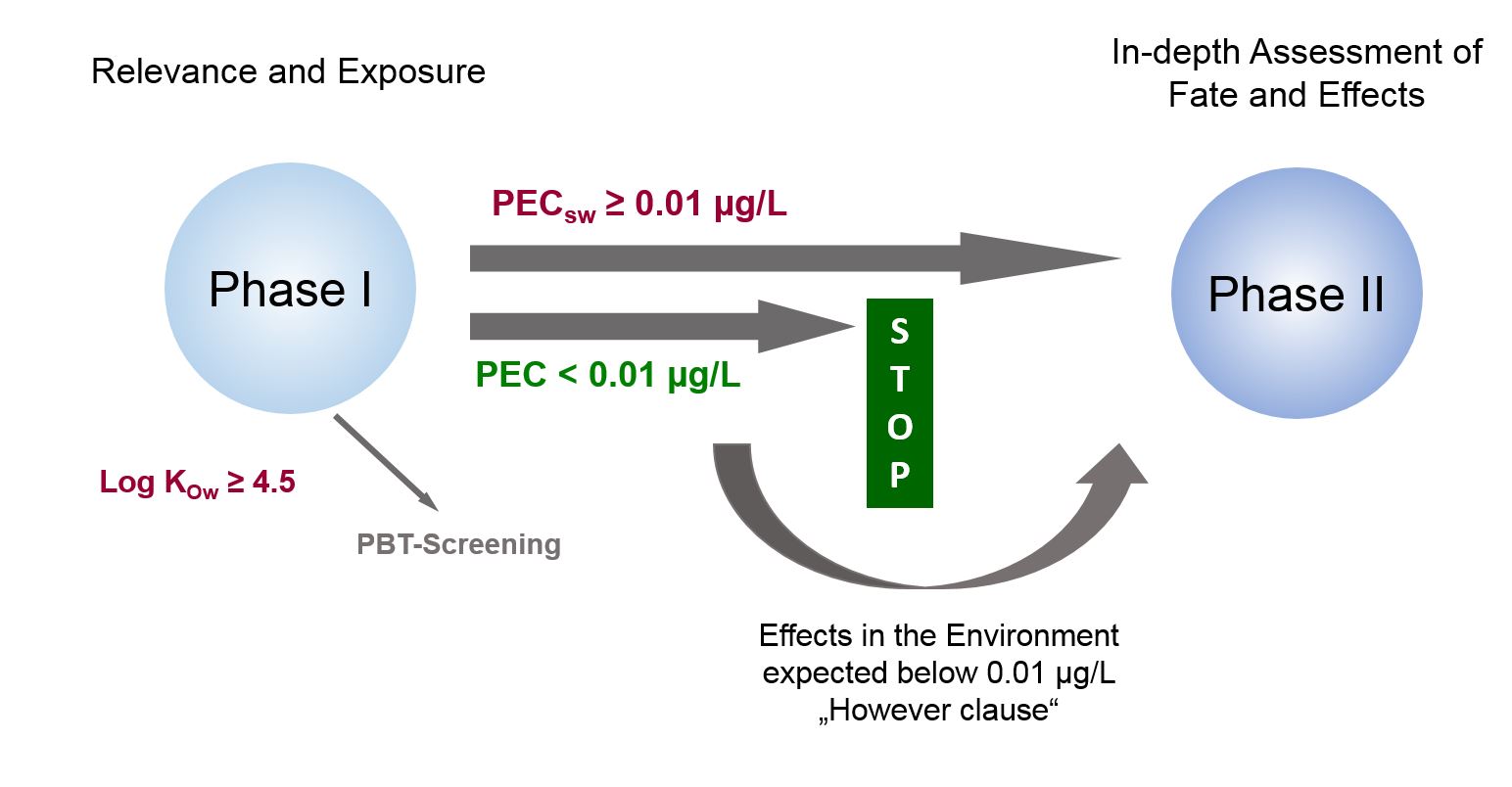


 Cornelis A.M. (Kees) van Gestel is retired professor of Ecotoxicology of Soil Ecosystems at the Vrije Universiteit Amsterdam. He has been working on different aspects of soil ecotoxicology, including toxicity test development, bioavailability, mixture toxicity, toxicokinetics, multigeneration effects and ecosystem level effects. His main interest is in linking bioavailability and ecological effects.
Cornelis A.M. (Kees) van Gestel is retired professor of Ecotoxicology of Soil Ecosystems at the Vrije Universiteit Amsterdam. He has been working on different aspects of soil ecotoxicology, including toxicity test development, bioavailability, mixture toxicity, toxicokinetics, multigeneration effects and ecosystem level effects. His main interest is in linking bioavailability and ecological effects. Frank G.A.J. Van Belleghem is an associate professor of environmental toxicology at the Open University of the Netherlands and researcher at the University of Hasselt (Belgium). His teaching activities are in the field of biology, biochemistry, (environmental) toxicology and environmental sciences. His research covers the toxicity of environmental pollutants including heavy metals, nanoparticles & microplastics and assessing (ultrastructural) stress with electron- and fluorescence microscopy.
Frank G.A.J. Van Belleghem is an associate professor of environmental toxicology at the Open University of the Netherlands and researcher at the University of Hasselt (Belgium). His teaching activities are in the field of biology, biochemistry, (environmental) toxicology and environmental sciences. His research covers the toxicity of environmental pollutants including heavy metals, nanoparticles & microplastics and assessing (ultrastructural) stress with electron- and fluorescence microscopy. Nico W. van den Brink is professor Environmental Toxicology at Wageningen University. His main research focus is on effects of chemicals on wildlife, especially under long term, chronic exposures. Furthermore, he has a great interest in polar ecotoxicology, both in the Antarctic as Arctic region.
Nico W. van den Brink is professor Environmental Toxicology at Wageningen University. His main research focus is on effects of chemicals on wildlife, especially under long term, chronic exposures. Furthermore, he has a great interest in polar ecotoxicology, both in the Antarctic as Arctic region. Steven T.J. Droge is senior researcher at Wageningen Environmental Reseach. He earlier worked at the Dutch Pesticide Registration Authority (CTGb) and as lecturer Environmental Chemistry at the University of Amsterdam. He worked on measuring and understanding bioavailability in toxicity studies of organic chemicals, ionogenic compounds in particular, during projects at Vrije Universiteit Amsterdam, Utrecht University, and Helmholtz Centre for Environmental Research - UFZ Leipzig. The most fascinating chemical he worked with was scopolamine.
Steven T.J. Droge is senior researcher at Wageningen Environmental Reseach. He earlier worked at the Dutch Pesticide Registration Authority (CTGb) and as lecturer Environmental Chemistry at the University of Amsterdam. He worked on measuring and understanding bioavailability in toxicity studies of organic chemicals, ionogenic compounds in particular, during projects at Vrije Universiteit Amsterdam, Utrecht University, and Helmholtz Centre for Environmental Research - UFZ Leipzig. The most fascinating chemical he worked with was scopolamine. Timo Hamers is an associate professor in Environmental Toxicology at the Vrije Universiteit Amsterdam. His main interest is in the application, development and optimization of small-scale in vitro bioassays to determine toxicity profiles of sets of individual compounds and complex environmental mixtures of pollutants as found in abiotic and biotic environmental and human samples.
Timo Hamers is an associate professor in Environmental Toxicology at the Vrije Universiteit Amsterdam. His main interest is in the application, development and optimization of small-scale in vitro bioassays to determine toxicity profiles of sets of individual compounds and complex environmental mixtures of pollutants as found in abiotic and biotic environmental and human samples. Joop L.M. Hermens is a retired associate professor of Environmental Toxicology and Chemistry at the Institute for Risk Assessment Sciences (IRAS) of Utrecht University. His research was focused on understanding the exposure and bioavailability of contaminants in the environment in relation to effects on ecosystems and on human health. Main topics also included the development of predictive methods in ecotoxicology of organic contaminants and mixtures.
Joop L.M. Hermens is a retired associate professor of Environmental Toxicology and Chemistry at the Institute for Risk Assessment Sciences (IRAS) of Utrecht University. His research was focused on understanding the exposure and bioavailability of contaminants in the environment in relation to effects on ecosystems and on human health. Main topics also included the development of predictive methods in ecotoxicology of organic contaminants and mixtures. Michiel H.S. Kraak is professor of Aquatic Ecotoxicology at the University of Amsterdam, where he started his academic career in 1987. He has published >120 papers in peer reviewed journals. Michiel Kraak has been supervising more than 20 PhD students and many more undergraduate students.
Michiel H.S. Kraak is professor of Aquatic Ecotoxicology at the University of Amsterdam, where he started his academic career in 1987. He has published >120 papers in peer reviewed journals. Michiel Kraak has been supervising more than 20 PhD students and many more undergraduate students. Ansje J. Löhr is associate professor of Environmental Natural Sciences at the Open University of the Netherlands. She has a background in marine biology and ecotoxicology. Her main research topic is on marine litter pollution on which she works in varying international interdisciplinary teams. She is working with UN Environment on worldwide educational and training programs on marine litter - activities of the Global Partnership on Marine Litter (GPML) - focusing on both monitoring and assessment, and reduction and prevention of marine litter pollution.
Ansje J. Löhr is associate professor of Environmental Natural Sciences at the Open University of the Netherlands. She has a background in marine biology and ecotoxicology. Her main research topic is on marine litter pollution on which she works in varying international interdisciplinary teams. She is working with UN Environment on worldwide educational and training programs on marine litter - activities of the Global Partnership on Marine Litter (GPML) - focusing on both monitoring and assessment, and reduction and prevention of marine litter pollution. John R. Parsons was a retired assistant professor of Environmental Chemistry at the University of Amsterdam. His main research interest was the environmental fate of organic chemicals and in particular their degradation by microorganisms and how this is affected by their bioavailability. He was also interested in how scientific research can be applied to improve chemical risk assessment. John passed away on 12 November 2024.
John R. Parsons was a retired assistant professor of Environmental Chemistry at the University of Amsterdam. His main research interest was the environmental fate of organic chemicals and in particular their degradation by microorganisms and how this is affected by their bioavailability. He was also interested in how scientific research can be applied to improve chemical risk assessment. John passed away on 12 November 2024. Ad M.J. Ragas is professor of Environmental Natural Sciences at the Open University of the Netherlands in Heerlen. He furthermore works as an associate professor at the Radboud University in Nijmegen. His main expertise is the modelling of human and ecological risks of chemicals. He covers topics like pharmaceuticals in the environment, (micro)plastics and the role of uncertainty in model predictions and decision-making.
Ad M.J. Ragas is professor of Environmental Natural Sciences at the Open University of the Netherlands in Heerlen. He furthermore works as an associate professor at the Radboud University in Nijmegen. His main expertise is the modelling of human and ecological risks of chemicals. He covers topics like pharmaceuticals in the environment, (micro)plastics and the role of uncertainty in model predictions and decision-making. Nico M. van Straalen is a retired professor of Animal Ecology at Vrije Universiteit Amsterdam where he was teaching evolutionary biology, zoology, molecular ecology and environmental toxicology. His contributions to ecotoxicology concern the development of statistical models for risk assessment and the application of genomics tools to assess toxicity and evolution of adaptation.
Nico M. van Straalen is a retired professor of Animal Ecology at Vrije Universiteit Amsterdam where he was teaching evolutionary biology, zoology, molecular ecology and environmental toxicology. His contributions to ecotoxicology concern the development of statistical models for risk assessment and the application of genomics tools to assess toxicity and evolution of adaptation. Martina G. Vijver is professor of Ecotoxicology at Leiden University since 2017. Her focus is on obtaining realistic predictions and measurements of how existing and emerging chemical stressors potentially affect our natural environment and the organisms living therein. She is specially interested in field realistic testing, and works on pesticides, metals, microplastics and nanomaterials. She loves earthworms, isopods, Daphnia and zebrafish larvae.
Martina G. Vijver is professor of Ecotoxicology at Leiden University since 2017. Her focus is on obtaining realistic predictions and measurements of how existing and emerging chemical stressors potentially affect our natural environment and the organisms living therein. She is specially interested in field realistic testing, and works on pesticides, metals, microplastics and nanomaterials. She loves earthworms, isopods, Daphnia and zebrafish larvae.{kind=link}
{kind=link}
{kind=link}
The arrangement Environmental Toxicology, an open online textbook is made with Wikiwijs of Kennisnet. Wikiwijs is an educational platform where you can find, create and share learning materials.
- Author
- Last modified
- 25-11-2025 15:17:19
- License
-


This learning material is published under the Creative Commons Attribution 4.0 International license. This means that, as long as you give attribution, you are free to:
- Share - copy and redistribute the material in any medium or format
- Adapt - remix, transform, and build upon the material
- for any purpose, including commercial purposes.
More information about the CC Naamsvermelding 4.0 Internationale licentie.
Additional information about this learning material
The following additional information is available about this learning material:
- Description
- This open online textbook aims at providing the student with basic knowledge on different aspects of Environmental chemistry, Environmental toxicology, Ecotoxicology and Ecotoxicological risk assessment. The book has modular design, each module having some clear learning objectives, a limited amount of information and some questions to check whether the knowledge offered is understood.
- End user
- leerling/student
- Difficulty
- gemiddeld
- Learning time
- 4 hour 0 minutes
- Keywords
- ecotoxicological risk assessment, ecotoxicology, environmental chemistry
Sources
| Source | Type |
|---|---|
|
Developments in Environmental Toxicology: interview with two pioneers https://youtu.be/08owH-YMuoI |
Video |
Used Wikiwijs arrangements
Toxicologie tekstboek SURF. (z.d.).
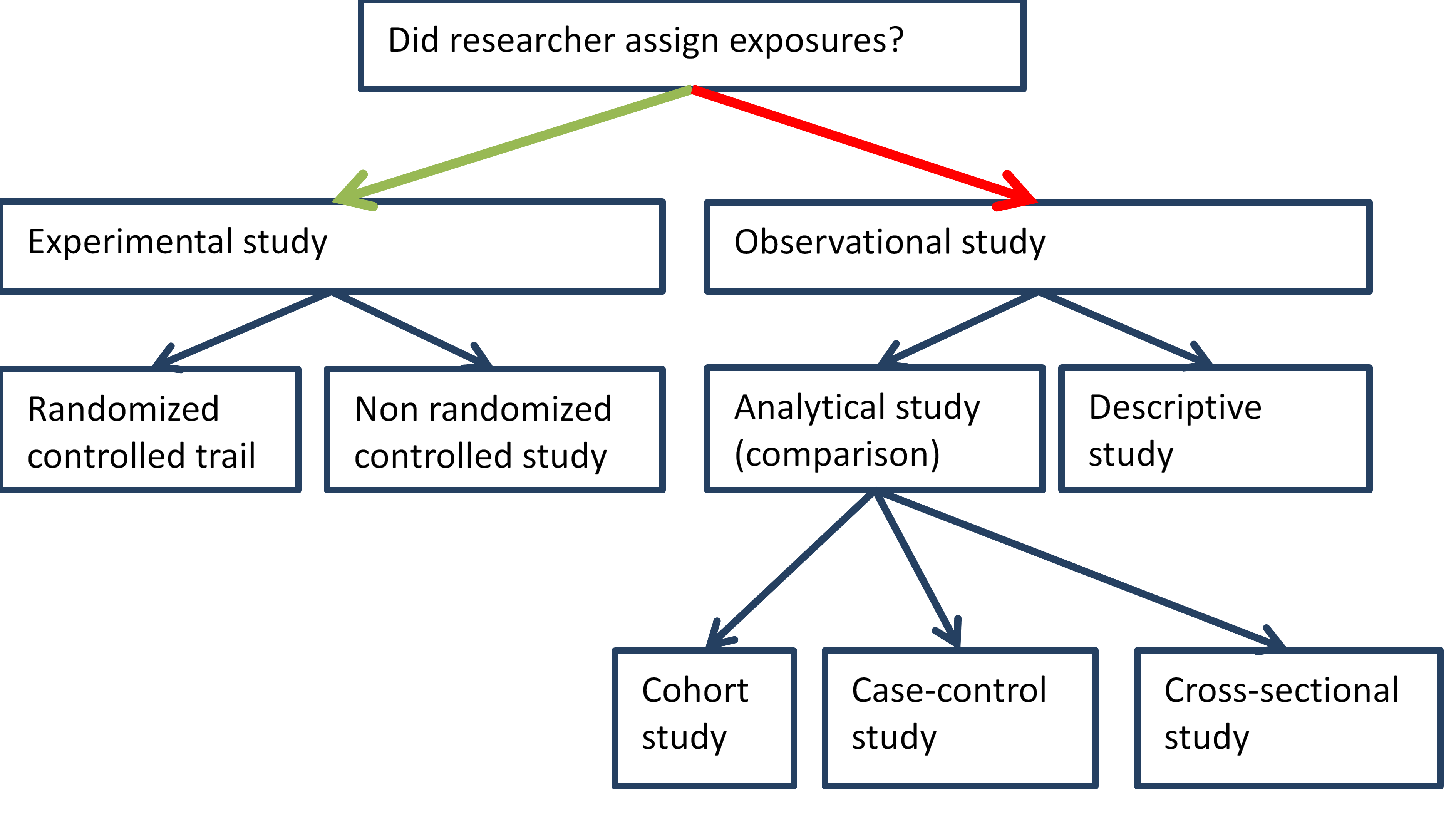
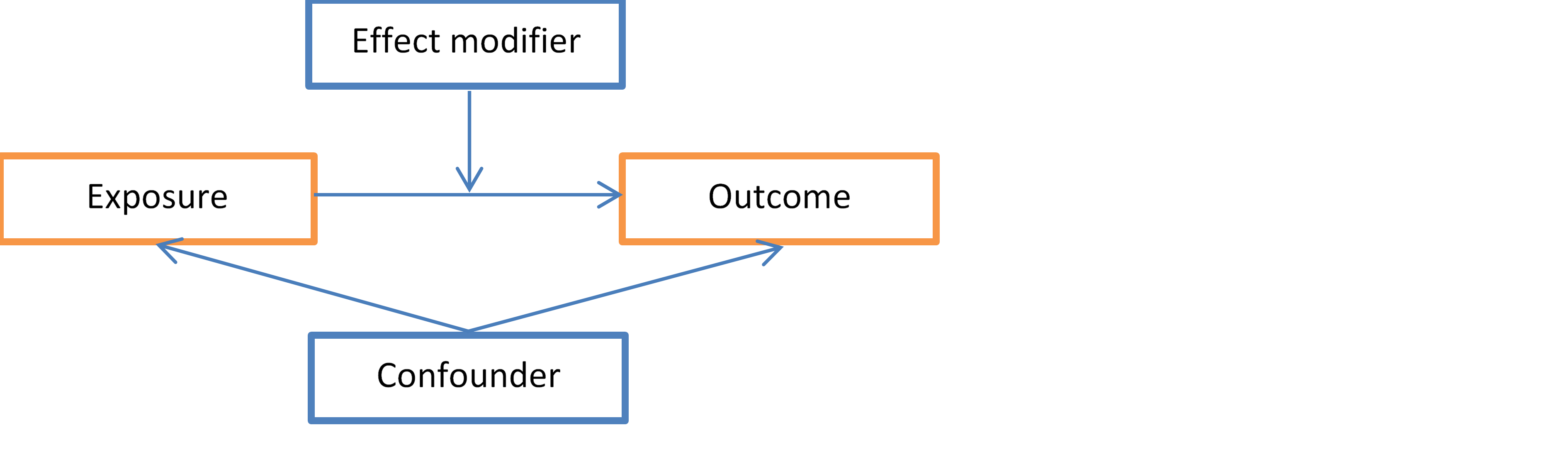
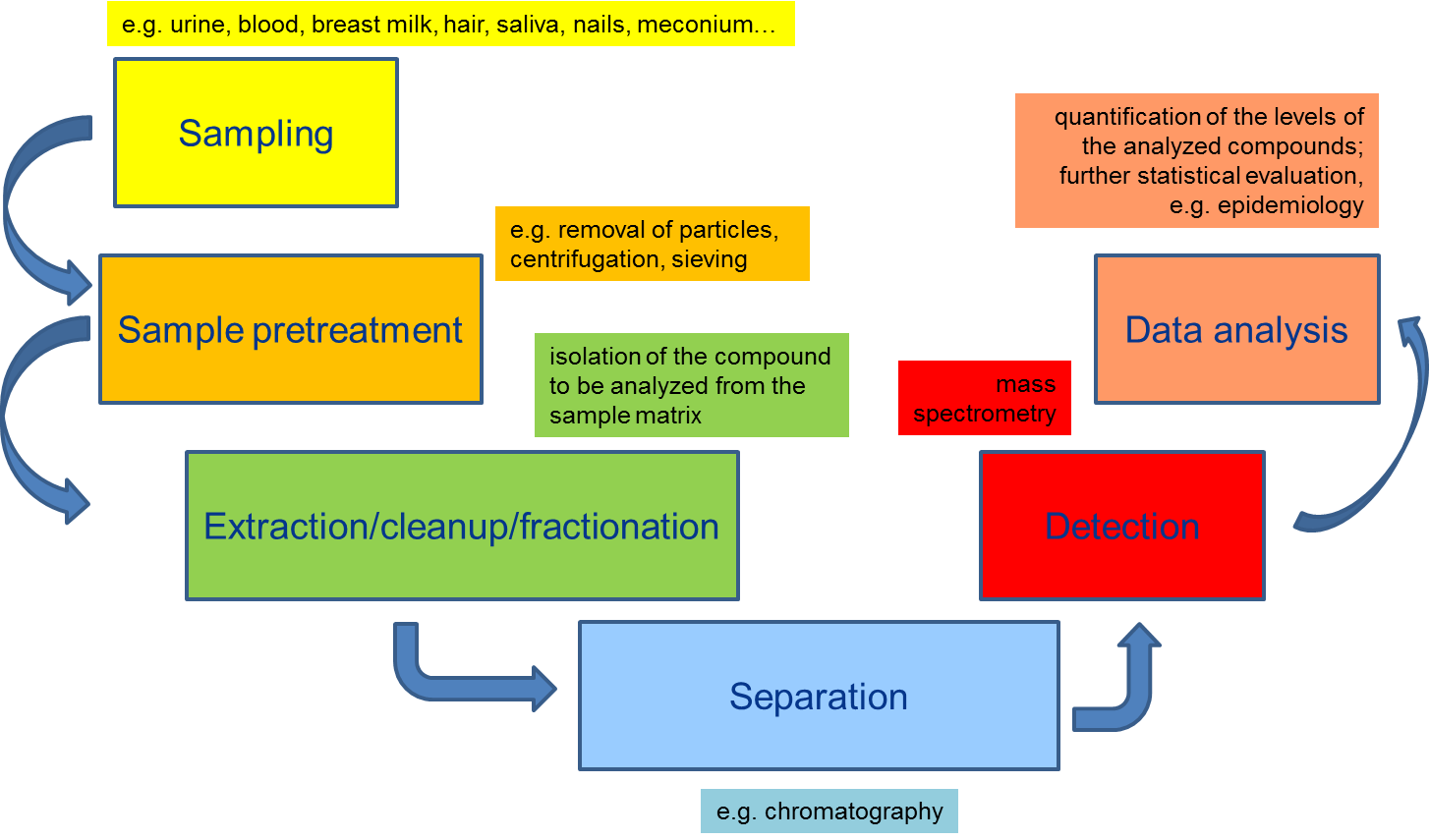
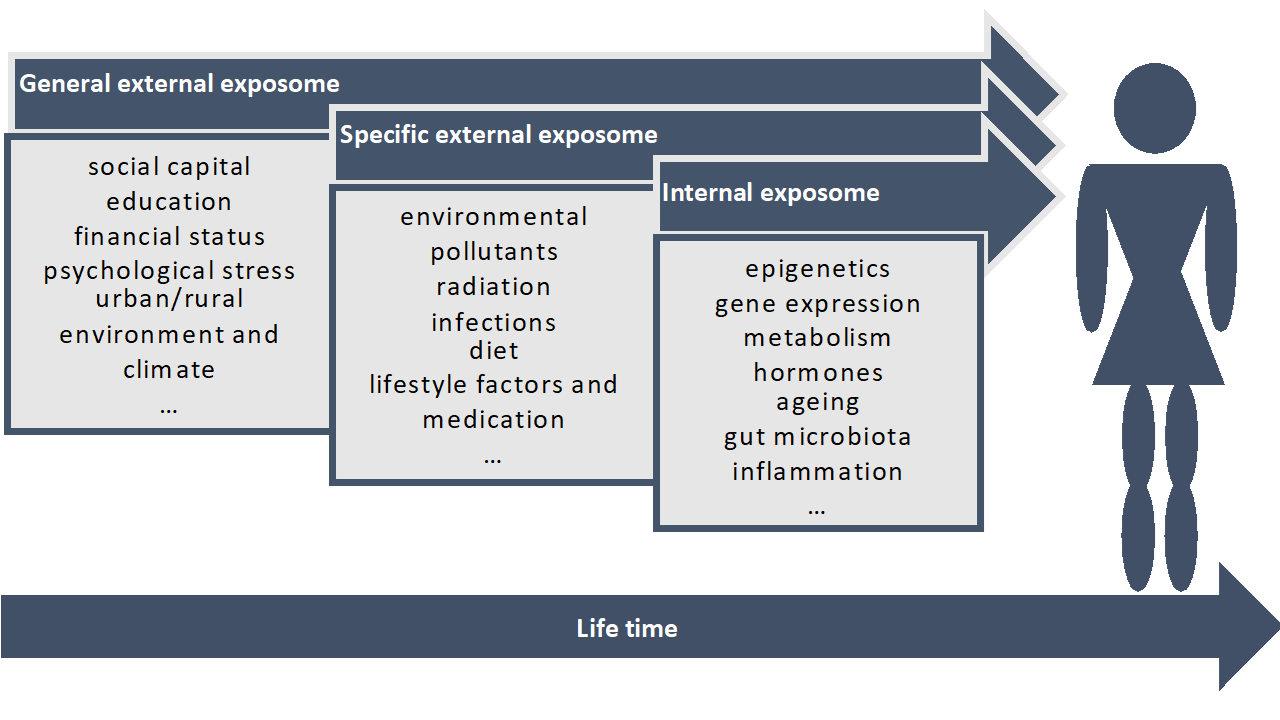
1. Introduction
Toxicologie tekstboek SURF. (z.d.).
2. Environmental Chemistry - Chemicals
https://maken.wikiwijs.nl/120172/2__Environmental_Chemistry___Chemicals
Toxicologie tekstboek SURF. (z.d.).
3. Environmental Chemistry - From Fate to Exposure
https://maken.wikiwijs.nl/120175/3__Environmental_Chemistry___From_Fate_to_Exposure
Toxicologie tekstboek SURF. (z.d.).
5. Population, Community and Ecosystem Ecotoxicology
https://maken.wikiwijs.nl/120181/5__Population__Community_and_Ecosystem_Ecotoxicology
Toxicologie tekstboek SURF. (z.d.).
6. Risk Assessment & Regulation
https://maken.wikiwijs.nl/120183/6__Risk_Assessment___Regulation
Toxicologie tekstboek SURF. (z.d.).
7. About this textbook